
Cushing’s Syndrome (CS) in Children: Pathophysiology, Clinical Features, Diagnostic, and Therapeutic Strategies
Sana Ashraf1* , Shakeela Parveen1,2 , Saba Ashraf1, Uzma Batool1, Mehwish Sultana1, Urwah Ishaque1 , Zainab Shafqat1 , Saman Shabbir1 ,Zainab Riaz1 ,and Zunaira Faiz1
1Department of Zoology, The Government Sadiq College Women, University of Bahawalpur, Pakistan
2Department of Zoology, University of Agriculture, Faisalabad, Pakistan
ABSTRACT
A rare condition known as Cushing’s Disease (CD) causes increased morbidity or mortality. It is an endocrine illness that presents differently from other endocrine disorders, making it difficult for doctors to control. Hence, early detection and treatment are essential. Significant fetal and mental complications during pregnancy are linked to Cushing’s Syndrome (CS). Except in the late trimester, surgery is its preferred method of treatment during pregnancy, with medication therapy as a backup. Therapeutic methods such as anti-cortisol medication, bilateral adrenalectomy, or radiation procedures may, therefore, be required to prevent long-term dangers of hypercortisolism, such as hirsutism, moon face, facial plethora, and obesity. Endogenous hypercortisolism increases the risk of cardiovascular metabolic symptoms, osteoporosis, respiratory diseases, psychological difficulties, and infections, while also inducing a high rate of morbidity or mortality.
Fig 1. Graphical Abstract
Keywords: clinical features, Cushing’s Syndrome (CS), diagnostic features, hypercortisolism, pathophysiology, therapeutic strategies
Cushing’s Syndrome (CS) is a collection of signs and symptoms generated by a permanent increase in the circulating free cortisol [1]. There is a male predominance among patients in early childhood, while in later childhood there is a predominance of female patients, which decreases with age [2]. CS is caused by persistent exposure to disproportionately increased levels of exogenous and endogenous cortisol [3]. Exogenous and endogenous are its two types having the same indications. The only difference is in the way they are launched. Exogenous CS is the most common and occurs in patients taking cortisol-like drugs, such as prednisone [4]. Endogenous CS is rare and difficult to diagnose. It is caused by Adrenocorticotropin-independent hypercortisolemia Hormone (ACTH) (adrenal) or ACTH-dependent sources (pituitary or ectopic) [5]. Its diagnosis is often difficult and demands repeated blood, urine, and saliva tests to detect the higher levels of cortisol caused by either a pituitary or an adrenal problem [6]. This condition is often known as Cushing’s Disease (CD) when tumors form in the pituitary gland that deregulate the secretions of ACTH and it may make a person prone to CS [7]. The most common cause is the use of iatrogenic corticosteroids with cushingoid features. The levels of circulating corticosteroids can also be increased by herbal preparations and other supplements in CS [8]. Hypercortisolism is interchangeable with CS. CD is an ACTH-dependent excess of cortisol due to a pituitary adenoma [9] and it is responsible for 80% of endogenous CS [10]. In patients with CS, impaired glucose metabolism contributes to significantly higher cardiovascular morbidity and mortality. Glucose metabolism should be subsequently monitored and properly targeted for glycemic control as in diabetic patients, even though their fasting glucose levels remain normal [11]. Keeping in view the importance of this problem, this study sought to investigate and provide insight into the pathophysiology and treatment options for CS (reduction of corticosteroids, surgery, radiation therapy and drugs.
1.1. Pathophysiology
Cortisol is a steroid hormone produced by fasciculus muscles in the adrenal cortex. Cortisol-binding proteins transport it to various body parts. These proteins bind nearly 90% of cortisol and its bioavailability ranges from 60% to 100%. The pathophysiology of CS and hypertension is quite complex [12]. The mechanisms that lead to hypertension are intricate and only partially comprehended. The bioavailability and potency of synthetic corticosteroids vary but they all have similar effects on signaling pathways. It is a catabolic hormone released during trauma. Insulin resistance is exacerbated and gluconeogenesis and glycogenolysis rates are sped up by excess cortisol. It has a direct impact on protein transcription and translation, on enzymes involved in protein synthesis, fat metabolism, glycogen, and the Krebs cycle. This hormone encourages the body to produce free glucose and causes an increase in glucose levels and insulin resistance. Gluconeogenesis makes the use of amino acids obtained from the breakdown of proteins. Osteoporosis, poor wound healing, and purple striae on the trunk are all consequences of prolonged protein catabolism. Collagen is one of the three amino-based proteins involved in all of these processes [13].
High cortisol levels also contribute to immune disorders. The number of lymphocytes decreases and the number of neutrophils rises as a result of this hormone. It raises neutrophil levels in the bloodstream but it does not increase neutrophil production. The regulation of AMP kinase, superoxide dismutase, glycogen phosphorylase, and numerous other enzymes is reversible by corticosteroids. The production of IFN-alpha, TNF-alpha, IL-2, and gamma is stymied by cortisol lymphocyte proliferation which can be stopped by lowering IL-2 levels [14]. As a result, significant effects may also be motivated by additional androgen secretion, despite the fact that atypical cortisol levels may be responsible for the majority of abnormalities caused by CS. Hypercortisolism can develop for a variety of reasons, such as an adenoma of the anterior pituitary gland that produces a lot of ACTHS and causes adrenal hyperplasia and excess cortisol production. Hind limb of hypothalamus is responsible to produce too much corticotropin-releasing hormone and causes too much of ACTH. CD occurs when the anterior pituitary secretes additional ACTH as a result of CS.
Excessive ACTH secretion, characterized by elevated plasma levels of ACTH and cortisol, is the most common cause of CS. Due to the inhibition of cortisol secretion by the anterior pituitary gland, approximately 20% to 25% of CS cases are caused by primary adrenal overproduction of cortisol, which has been linked to decreased ACTH levels.
1.2. Etiology
Endogenous and exogenous cortisol hypercortisolism are the two primary drivers of CS. Endogenous CS is caused by excess cortisol production by adrenal glands [15], which can be ACTH-dependent or ACTH-independent. According to [16], ectopic ACTH secretion by ACTH-secreting pituitary adenomas and neoplasms is the cause of ACTH-dependent CS. Adrenal hyperplasia, adenoma, and carcinoma are the main causes of ACTH-independent CS [17].
Table 1. Etiology of Cushing’s Syndrome (CS)
|
ACTH-dependent CS |
ACTH-independent CS |
|
1. Administration of CS (ACTH-secreting pituitary adenoma) |
1. Exogenous glucocorticoid |
|
2. Ectopic ACTH syndrome |
2. Adrenocortical tumor (adenoma or carcinoma) |
|
|
3. Primary adrenocortical hyperplasia - macronodular adrenal hyperplasia -, Carney Complex/MEN PPNAD, - McCune-Albright syndrome |
1.3. Clinical Features
Age, duration, and intensity of hypercortisolism in patients affect the clinical indices of CS. Most children experience an incremental development of CS [5]. A poor lifestyle, sickness, and five times the normal mortality rate are all associated with CS. Patients may exhibit higher Body Mass Index (BMI) with the deposition of fat in dorsocervical and supraclavicular regions, proximal muscular weakness, hypertension, hirsutism, glucose intolerance, exhaustion, thin skin with prominent purple stride, and irregular menstruation in severe cases [18]. CS is highly prevalent among patients with diabetes mellitus [19]. Skin changes, weakness, obesity, hypertension, osteoporosis, and psychological issues such as depression, dyslipidemia, irregular menstruation, glucose intolerance, impotence, and growth retardation among children are all symptoms of CS [20]. Based on this clinical presentation, CS can be identified and treated.
Table 2. Clinical Features
|
Clinical Features |
Citation |
|
The risk of developing CS is significant in people with diabetes mellitus. |
[6] |
|
Excessive facial hair, purple stride, buffalo hump, truncal obesity, and supraclavicular fat pads. |
[21] |
|
Poor quality of life, morbidity, 5-fold excess mortality, central obesity with fat deposits in the dorsocervical and supraclavicular regions, thin skin with prominent purple stride, exhaustion, proximal muscle weakness, hypertension, glucose intolerance, acne, hirsutism, and irregular menstrual cycles. |
[18] |
|
Additionally, Carney Complex (CNC), a rare multiorgan tumoral condition with a dominant hereditary pattern, is connected to CS. |
[22] |
|
Obesity, hypertension, skin changes, osteopenia, weakness, and psychological issues such as depression, irregular menstruation, glucose intolerance, impotence, dyslipidemia, and stunted growth in children are all symptoms of CS. |
[20] |
1.4. Diagnosis
The confirmation of hypercortisolism is the first step in the diagnostic evaluation of CS. The section on laboratory testing describes the confirmation process in more detail. Once the existence of endogenous hypercortisolism has been validated, there are two main diagnostic methods that can be used to identify the cause of excessive cortisol production. The initial strategy is to distinguish between CS etiologies that depend on ACTH and those that do not. The second method involves determining the age-based etiology of CS. Infancy McCune-Albright Syndrome (MAS) is the condition with which endogenous CS is most frequently linked. Before the age of four, adrenocortical tumors are the most prevalent cause. Cushing Disease (CD) [1], beyond the age of five, is most frequently caused by an adenoma of the pituitary gland that secretes ACTH [1]. Males are more likely than females to have CD as prepubescent children [23].
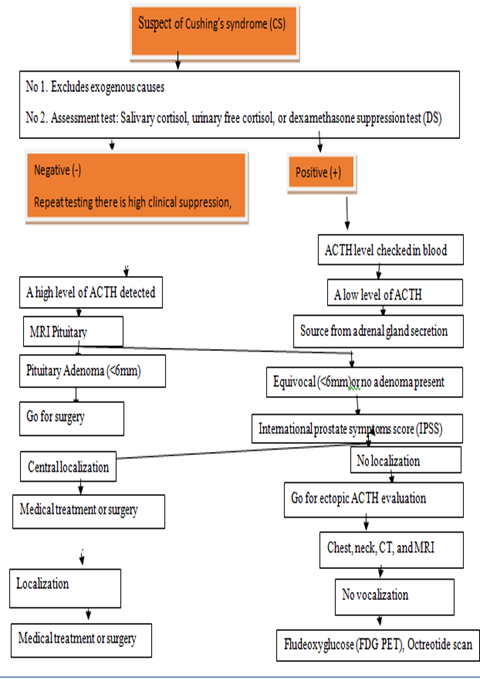
Fig 2. Diagnosis of CS (Algorithm for the diagnosis and management of CS)
2.1. Assessment for CS starts with the confirmation of hypercortisolism
(24 hour) Urinary Free Cortisol Excretion (UFC)
Urinary Free Cortisol (UFC) excretion on 3 consecutive days is regarded by many endocrinologists as the first-line test. It is characterized by low specificity but great sensitivity [24]. In children, UFC should be adjusted for body surface area / 1.72 m2. The average amount is less than 70 mcg/m2/d. If the levels are above the normal level, further analysis becomes necessary. Values more than four times the average value are regarded as diagnostic [25].
2.2. Midnight Plasma and Salivary Cortisol Level
Since minimum cortisol production occurs during night time, these tests benefit from the circadian cycle of cortisol secretion. To prevent a cortisol response to venipuncture, children must sleep with an intravenous catheter for the overnight plasma cortisol test. A late night plasma cortisol level of ≥4.4μg/dL was determined in an NIH retrospective assessment to be 100% specific and 99% sensitive for confirming the diagnosis of CS [26].
Salivary levels of cortisol are used for the first diagnosis of hypercortisolism, with the advantage of home saliva collection. In patients having CD, the circadian salivary cortisol cycle is missing [27]. In a study of children, the clinical efficacy of overnight salivary cortisol and UFC m2 of the body’s surface area were found to be the same (93%) [28].
2.3. Low Dose Dexamethasone Suppression Test
There are many different protocols. Typically, for kids who weigh under 40 kg, 0.5mg of oral dexamethasone is administered every 6 hour for a total of 8 doses (or 30µg/kg/d for 2 days). Serum cortisol levels are evaluated at baseline, 48 hours later, and 6 hours after the final dosage. In healthy people, the level of cortisol should be minimal (< 50 nmol/L, < 1.8µg/dL). For the identification of CS and the exclusion of diagnosis, this test has a sensitivity of 97% and a specificity of close to 100% [29].
2.4. Overnight 1mg Dexamethasone Suppression Test
At 11 pm, 1mg of oral dexamethasone is given and blood cortisol levels are measured at 8 am the following morning. In individuals with CS, the setpoint for ACTH production is above normal and the levels of cortisol are not suppressed, even following a mild dosage of dexamethasone. In this test, the plasma cortisol level setting at 8 am is less than 2µg/dL (50 nmol/L) in normal patients. If the level is higher than 10µg/dL (> 275 nmol/L), then the cortisol concentration suggests the diagnosis of CS. Whereas, the significance of intermediate levels regarding diagnosis remains unclear. This test has a limited sensitivity of just 55% and there is a significant percentage of inaccurate results (15–20%) [30].
Difference between ACTH-independent and ACTH-dependent causes is inspected, once hypercortisolism is confirmed.
2.5. Plasma ACTH
All of the 45 children with pediatric CS who had adrenal causes of hypercortisolism were found to have undetectable plasma ACTH levels. There were measurable amounts of ACTH in all CD patients, ranging from 12 to 128 ng/L [31]. Adrenal imaging should be stimulated by an undetectable ACTH level. A midnight plasma ACTH reveals an excess of ACTH because the level is more than 23 ng/L (5 pmol/L) [32].
2.6. Overnight 8mg Dexamethasone Suppression Test
Ectopic ACTH secretion, a pituitary adenoma, is caused by excessive Corticotropin-Releasing Hormone (CRH) production by hypothalamus (very uncommon). The inhibition of ACTH production by pituitary adenomas and glucocorticoids are the symptoms for excessive ACTH production. At 11 pm, a baseline plasma cortisol measurement and 8 mg of oral dexamethasone are given and a subsequent plasma cortisol measurement is taken at 8 am the following day. In contrast to ectopic ACTH production, a reduction in plasma cortisol of at least ≥ 50 % is thought to be diagnostic of CD [33].
2.7. Corticotropin-Releasing Hormone (CRH) Test
Synthetic forms of human or ovine CRH are used in the CRH test. The CRH test results in elevated cortisol and ACTH levels in the majority of pituitary tumors. If the post-CRH ACTH concentration is greater than 34% of the baseline assessment or the cortisol level is greater than 20% of the baseline value, the test is deemed positive for CS [34]. However, ectopic ACTH-producing tumors can also respond to CRH and the outcomes are inconsistent when separating CD from EAS.
In 2015, the endocrine society issued guidelines to treat CS, although they were not specifically for children [35]. Transsphenoidal surgical resection of the pituitary mass secreting ACTH is the treatment of choice for this condition [36]. Treatments for CS include the following.
3.1. Reducing the Amount of Corticosteroid
Symptoms of CD due to taking corticosteroid drugs for a prolonged period of time can be controlled by reducing the dose of the prescribed drugs over time, following the suggestion of the physician. If medicine is stopped suddenly, cortisol levels drop. If cortisol declines slowly, the body can resume normal cortisol production [37].
3.2. Surgery
The appropriate therapy for hypercortisolism is “surgical resection” and adrenal tumors, extopic tumors or CS may be the leading causes of this resection [38]. Transsphenoidal surgical resection is an initial therapeutic intervention to treat CD [26]. Depending on the neurosurgeon’s experience and local invasion, 50-70% of macroadenomas and 60-90% of microadenomas can be operated on. Remission is defined by normal circadian ACTH and cortisol rhythms and cortisol is suppressed by an overnight/low-dose dexamethasone suppression test. However, it remains difficult to predict which patients are at a high risk of recurrence, as some patients who do not heal quickly after surgery may have delayed remission. It is also difficult to evaluate the cured patients previously suffering with CD because of the high risk of recurrence. In other words, even long-term postoperative remission requires at least a long-term close clinical follow-up. Therapies are also available when immediate surgical failure occurs, such as a second pituitary surgery, therapy, radiation technique, or bilateral adrenalectomy. These are the only treatments that can lead to a long-term reduction of the disease [39].
3.3. Radiation Therapy
If the whole surgical resection of the pituitary adenoma isn’t always viable or if the affected person refuses surgery, radiation remedy gives the capacity for pituitary remission [40]. Recently, various approaches to Stereotactic Radiosurgery (SRS) have been used (gamma knife, linear accelerator, proton beam). For radiotherapy, the ideal dose for SRS is approximately 20-25 Gy, with 45-50.4 Gy given over 5 weeks [41]. Loss of pituitary function is a major risk of fractionated radiation therapy and is probably the obvious consequence of radiation and surgery. In about 20-40% of patients, there is a loss of pituitary function (about) 10 years after radiotherapy and this risk increases subsequently [42].
3.4. Medication
Akin to radiotherapy, drug therapy supports surgical treatment and remains the preferred treatment. Drug therapy of the pituitary gland is recommended in addition to radiotherapy or if surgery or radiotherapy is contraindicated then drug therapy is suggested. Several classes of therapies exist that retard the steroidogenesis in the adrenal gland. All are available to assist with pre-operative responses that include hypercortisolism as well as pituitary tumors of insignificant size. These include ketoconazole, metoripon, mitotane, aminoglutethimide, as well as intimidate. In practice, the most commonly used is ketoconazole at doses of 400–1200 mg per day, with increasing doses and monitoring for side effects (gastrointestinal disturbances, gonadal dysfunction, and hepatocyte lysis), high blood pressure, faintness, ataxia, torpor, and rash. op`DDD or Mitotane is an adrenolytic drug used 6-12 grams/day [43].
Fig 3. Treatment Options
Table 3. Treatment options and their attributes
|
Treatment |
Attributes |
|
Radiation therapy |
Stereotactic surgery (20-25GY) Best Dose (40-50GY for 5 weeks) |
|
Reducing corticosteroids use |
Regular cortisol action |
|
Medication |
Ketocoazole (400-1200mg per day), Mitotane (6-12g per day), Metoriopon (1-4g per day) & Aminoglutethimide. |
|
Surgery |
Remission through normal circadian ACTH and cortisol rhythms as well as night suppressed cortisol levels, Surgery (Tansphenoidol). |
CS can be both endogenous and exogenous. There are numerous ways to treat CS. Today, surgery can be used to successfully treat the majority of CS patients, either curing them or managing them [39]. Patients should be sent to major facilities because CS diagnosis and treatment still present significant challenges [43]. CS may be present in Type 2 diabetics with poor control, which has important ramifications for screening this at-risk population. A good screening test that might work for this purpose is salivary cortisol. Many people continue to be dissatisfied with the results of this CS treatment. In this field, advanced developments are needed [44].
REFERENCES
* Corresponding author: sana.ashrafsci@gmail.com