Molecular Diagnostic Strategies Used for the Detection of Microbial Pathogens in Human Beings: A Review
Saba Mahboob, Kalsoom Tahir, Sikander Ali*, and Muhammad Nauman Aftab
Government College University Lahore, Pakistan
Abstract
Background: Viral diseases pose a serious health hazard to human population, worldwide. A perfect illustration of how a viral infection could pose a serious threat to public health and economic sectors is the current COVID-19 outbreak brought on by SARS-CoV-2 in 2019. Consequently, obtaining a prompt and accurate diagnosis is the first step in treating infections. For effective treatment, epidemic control, and prevention, early and precise identification of microbial presence in patient samples is essential.
Methods: This study lists some of the molecular and immunological diagnostic methods that can be used to find infections in human beings. Rapid viral detection in patient samples is possible by the use of molecular diagnostic techniques. These techniques are also reasonably cheap, quite sensitive, and very targeted. Infections in human beings have been detected and the epidemiology of these illnesses has been widely studied using immunologically based methods.
Results: In clinical samples, these methods can identify viral antigens or antiviral antibodies. Many commercially accessible molecular and immunological diagnostic kits make it easier to employ these techniques in most clinical laboratories around the world.
Conclusion: This review offers a new perspective on molecular techniques employed in the application of the clinical diagnostics of microbes.
Introduction
The healthcare system is highly dependent on diagnostic techniques. The use of these techniques is essential for the right treatment and decision-making in healthcare systems. At every stage, medical diagnostic techniques provide critical insights regarding treatment, detection, diagnosis, and better management of health conditions [1]. There are many conventional methods used in diagnostics for the diagnosis of diseases caused by pathogens. However, many pathogens are difficult to grow in the laboratory using conventional methods of cultivation. Studies have indicated that the cultivation ability of bacteria from natural samples is less than 1% [2]. There are many different factors affecting cultivation, such as lack of nutrients required for microbial growth, culture medium toxicity towards the desired organisms in a mixed population, production of harmful substances which inhibit the growth of desired microorganisms, and metabolic dependence of desired strain on other organisms for growth [3]. Many rapid immunological detection techniques can also be utilized for either direct identification of microorganisms or with the help of the immune responses of the host. In this scenario, enzyme assays such as enzyme-linked immune sorbent assay (ELISA) remains the most commonly used diagnostic technique. This technique is quick, cheap, and easy to standardize, although its sensitivity is very low, with almost 104 cells needed to produce results. The specificity of the process is varied and depends upon the antibody type used and the dead and live microorganisms [1]. Due to the underlying issues, rapid diagnostic techniques based on genetic material, such as RNA, DNA, and antibodies have been formulated for the identification of disease-causing microbes. Ideally, pathogen detection methods must be accurate, robust, less costly, sensitive, reproducible, and easy to operate. The hybridization methods for nucleic acids and polymerase chain reaction (PCR) assay exhibit these properties for the better identification of pathogens [4].
Molecular diagnostic techniques are advantageous over conventional diagnostic protocols as the former are highly insensitive, specific, and less costly for microbial identification, as compared to traditional methods. Better identification and discrimination of pathogens is carried out currently based on the high specificity of molecular diagnostic methods. These methods detect disease-causing microorganisms due to their high sensitivity directly from the clinical and environmental samples without the need for cultivation [5].
The term ''molecular disease'' was invented in 1949 by Pauling and his colleagues by observing diseases such as sickle cell anemia caused due to alternation in the β-globin chain. Their findings led to the foundations of molecular diagnostics, although during the course of years several big transformations have occurred in this field. New and advanced molecular diagnostic methods have revolutionized the field of practical clinical microbiology. There are many molecular diagnostic methods being used for the diagnosis of infections since the 1980s. Furthermore, Koch's postulates have been applied to identify the pathogenicity of bacteria at the gene level, instead of the whole organism level [6].
This brief review gives an understanding of advanced diagnostic techniques used for the identification of the pathogens in clinical microbiology. Molecular diagnostic methods used for the diagnosis of diseases might be categorized into numerous major groups [7]. These involve PCRbased techniques (such as traditional PCR, real-time PCR, RT-PCR, nested PCR, and broad range 16S rRNA gene PCR), RNA/DNA probe-based methods (including blotting techniques and direct hybridization techniques, such as in-situation hybridization, fluorescent in situ hybridization, PNA FISH), amplification-based techniques (included array-based techniques, loop-mediated isothermal amplification (LAP)) [1] sequence-based methods (included Sanger based bideoxy sequencing, pyrosequencing, multilocus sequencing typing (MLST), MALDI-TOF MS, nanopore technology), and other diagnostic methods for post-amplification [8]. Each molecular method has its advantages and limitations, illustrated in Table 1.
In this review article, advancements in molecular techniques used for the diagnosis of microbial pathogens in human beings are discussed, as shown in Figure 3. The focus remains on the emerging molecular techniques used for the identification of microbial pathogens to monitor human diseases.
Table 1. Advantages and Limitations of Molecular Techniques for the Diagnosis of Microbial Pathogens in Human Beings [9].
|
Techniques |
Advantages |
Limitations |
|
Conventional PCR |
Precise and rapid results when use specific primer |
Much cost and labor required |
|
RT-PCR |
Provide quantitative date, more subtle than traditional PCR |
Time consuming and costly |
|
Nested PCR |
Two primer sets used to increase the specificity and yield of amplified target DNA |
Due to two amplification cycles, risk of contamination occurs |
|
Real-time PCR |
Automated, no requirement of the post amplification analysis |
Complexity and cost due to instantaneous thermal cycling and the fluorescence detection |
|
Northern blotting |
For detection of the RNA size |
Used for small gene sample |
|
FISH |
Used for the non-dividing cells |
Difficult probe preparing method |
|
In-situ hybridization |
Extreme use of short-supply tissue |
Hard to identifying targets which have low RNA and DNA copies |
|
RNA-Seq |
Amplified sensitivity and specificity |
Costly, bioinformatics information necessary for the data analysis |
|
LAMP |
Sensitive, rapid, and specific |
Complex primer design, risk of contamination, recognize only definite pathogen |
|
Microarray
|
Used easily because it has not required large scale DNA sequencing |
Huge amount of the mRNA has been required |
2. CATEGORIZATION OF MOLECULAR TECHNIQUES
2.1. PCR-based Diagnostic Methods
The invention of polymerase chain reaction (PCR) has revolutionized the field of molecular diagnostics in terms of the identification, differentiation, and characterization of microorganisms, especially pathogens. PCR-based techniques have been highly specific and sensitive in their function and produce multiple copies from a single specific fragment of DNA. During the last 2 decades, many varieties of the standard PCR technique have been introduced. However, those mostly used for the identification of microbial pathogen detection are conventional PCR, nested PCR, real-time PCR, and broad range 16S rRNA gene PCR.
2.1.1. Conventional PCR. This amplification process runs with the help of specific primers via alternate cycles of denaturation, annealing, and elongation, as shown in Figure 1. This technique depends upon the efficiency of DNA extraction and concentration of deoxynucleoside triphosphate. At the beginning, PCR was only used for the detection of bacterial and viral diseases, although it is now used also for the detection of plant pathogens [10]. Sometimes, the function of PCR is compromised due to inhibitors present in the tested sample. However, PCR-specific primers designing is needed for the replication of DNA, which hinders its practical application in field sampling. Sometimes, a single primer pair is insufficient and produces faulty results. DNA primers and probes are widely used to limit this problem [11]. This is a much more specific technique but remains costly and needs a lot of labour in traditional PCR.
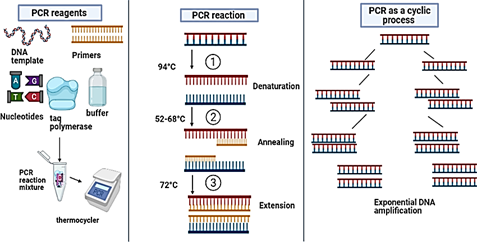
Figure 1. Overview of Conventional PCR [12]
2.1.2. Nested PCR. Nested PCR comprises two rounds of amplification. The first round is used to make copies of a large region of DNA via a single set of primers. Then, the product of the first round itself is used as a template for the second round by using two internal primers. Due to the two rounds of amplification, the cycle's sensitivity is enhanced, diluting any inhibitor present in the sample and increasing specificity. In the first round of amplification, if non-specific products are generated, then such products do not act as templates for the second round [13]. Due to an alternative round of amplification in separate tubes, there are chances of significant contamination. So, false positive results generated due to contamination and intense struggle are the main disadvantages of nested PCR [14]. Laboratories must take precautions and follow some advanced protocols to limit these drawbacks [15].
2.1.3. RT-PCR. Reverse transcription PCR was invented after the development of a large number of changes in traditional PCR. Today, RT-PCR is used in many medical and biological fields due to its diagnostic capabilities [16]. RT-PCR technique can identify the presence of any pathogens or nonpathogenic genetic material including viruses. In this molecular diagnostic technique, RNA is converted into complementary DNA (cDNA) reversely and then copies are made through the PCR technique with the help of random primers and reverse transcriptase enzyme. RT-PCR is widely used for the detection of infection-causing RNA viruses, such as Retroviruses. The diagnosis of RNA viruses is very useful for the development and checking of antimicrobial vaccine therapy [17]. Other conventional PCR could not distinguish between live and dead pathogens. However, with the discovery of RT-PCR, this drawback was eliminated. In fact, mRNA disintegrates in dead cells. So, mRNA is detected by RT-PCR for checking the viability of cells [18]. Fluorescent dyes, such as SYBR Green 1 or Taq Man probe, are used for the monitoring of reaction during the amplification process. The signal is produced when dye intercalates into the DNA. After each cycle of amplification, the amount of DNA increases with the increase of signal from the fluorescent dye [19]. RT-PCR is a less costly but a nonspecific technique. The disadvantage of this method is the formation of primer dimer when dye intercalates into the DNA. As a result, false positive results can be produced.
2.1.4. Real-time PCR. Real-time PCR techniques have been widely used in clinical microbiology. This method functions with the help of specific probes, which are labelled by fluorescent in addition to amplification techniques as shown in Figure 2. There are numerous different probes formats and instruments for the detection of pathogens are present, such as TaqMan hydrolysis probes, Scorpion probes, molecular beacons, and minor groove binding probes [20]. In this technique, the risk of contamination is minimized to a greater extent due to the closed tube system in real-time PCR. One of the major benefits of real-time PCR using fluorescence technology is that quantify genetic variations e.g., single nucleotide polymorphisms with the help of a set of precise probes for each possible SNP within the same species [21].
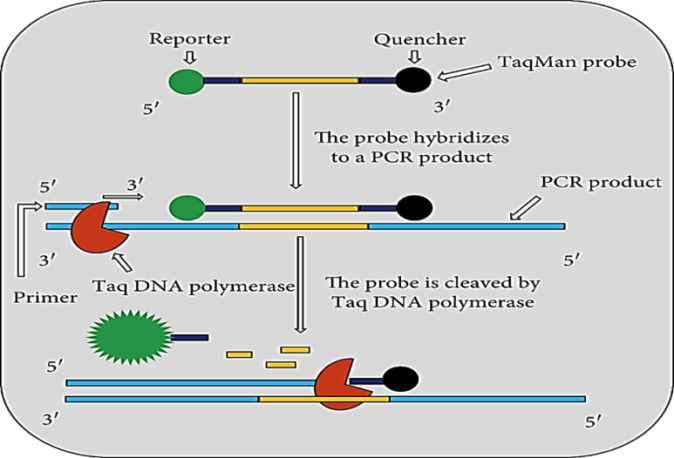
Figure 2. Schematic Representation of Real-time PCR [1]
2.1.5. Broad Range16s rRNA Gene PCR. Broad range 16S ribosomal ribonucleic acid gene polymerase chain reaction is used for pathogen detection and identification of clinical specimens [22]. This technique is usually applied for cases where bacterial culture gives negative results, despite suspected infection [23]. This technique is mainly used for fastidious bacteria that do not grow easily in the laboratory, such as Mycobacterium genovese, Coxiella burnetii, Ehrlichia chaffeensis, and Tropheryma whipplei [24]. Conserved regions of the 16S rRNA are ideal for designing primers, sequencing based on PCR, and alignment of sequences. Species identification is done by this gene because of having species-specific variable regions [25].
Universal primers have been utilized for the amplification of PCR products and gene sequencing of the 16S rRNA gene when a tested specimen is suspected to have nonculturable pathogens. Unknown bacteria were detected by comparing the gene sequence of 16S rRNA with the sequences already present in the database. Samples detected via PCR showed negative results specific for C. burnetii. However, when these samples were detected via 16S rRNA gene sequences, about 98% showed similarity to that of C. burnetii [26].
2.1.6. DiversiLab Repetitive Sequence-based PCR (DL rep-PCR). DiversiLab is a program based on a repetitive sequence PCR system for bacterial typing. It has a high degree of standardization, specifically at the electrophoresis step achieved through Bioanalyzer [27]. In contrast to conventional gel electrophoresis, this method via microfluidic capillary electrophoresis overwhelms the low duplicability of prior rep-PCR methodologies [28]. Also, it provides internet-based, user friendly, and computer supported data evaluation [29]. The consistency of data achieved through this system has been species-dependent [30].
2.1.7. Multiple Locus Variable Number Tandem Repeat Analysis (MLVA). It is a process (based upon PCR) that has been used to subtype the strains of microorganisms through the variable copy number of tandem repeats. MLVA applies to DNA sequences that differ in the number of tandem repeats obtained on multiple loci in bacterial genomes identified through PCR by utilizing flanking primers [31]. It is a more promising method as compared to other typing techniques due to greater diversity and comprehensive discrimination ability [32]. It is a rapid method with great resolution. MLVA microbial cells from the agar plate are taken and DNA is released by boiling the cells. For the detection of the DNA region, a variable number tandem repeat array is performed. PCR is used to combine DNA with chemicals for the amplification of VNTR. After PCR, the size of the PCR product must be determined. Then, the PCR product is loaded into the sample analysis plate and blended with chemicals which help to determine the product size. Capillary electrophoresis is used to determine the size of DNA fragments. Data outputs of MLA are expressed in the form of an electropherogram [33].
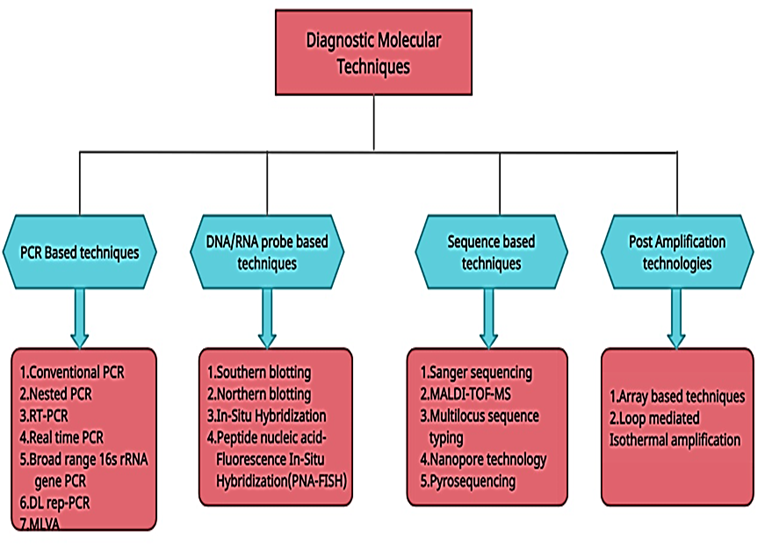
Figure 3. Overview of Molecular Techniques [9]
Table 2. Advantages and Limitations of PCR-based Techniques
|
Techniques |
Advantages |
Limitation |
References |
|
Conventional PCR |
· Widely employed · Specific and sensitive · Multiplex detection potential |
· Prone to inhibitors · Qualitative · Labour intensive · Time consuming · High risk of contamination |
[34] |
|
RT-PCR |
· Specific and sensitive · Multiplex detection potential |
· High risk of contamination · Difficult RNA handling · Time consuming · Prone to inhibitors · Expensive |
|
|
Real time PCR |
· Rapid highly specific and sensitive · Genotyping · Less labour intensive · Low cross contamination risk |
· Expensive laboratory equipment · Prone to inhibitor |
[38] |
2.2. DNA/RNA Probe-based Method (Sequencing Independent Method)
2.2.1. Southern Blotting. This technique is a detection method for the target DNA sequence. Firstly, restriction enzymes, such as restriction endonucleases, are used to cut the target DNA into short pieces. Then, gel electrophoresis technique is used to segregate these pieces. On gel, these fragments move according to their molecular weight. After gel electrophoresis, DNA fragments are shifted onto a membrane called the nitrocellulose membrane or blotting paper. These fragments are then incubated on blotting paper with selective probes. The probes are highly selective in their function and are radioactively labelled. Moreover, bind with the resolution 1 in a million fragments. In molecular biology, this technique is usually used for the detection of viral and bacterial infections. Autoradiography has been used for detection purposes after the completion of the incubation period, as shown in Figure 4 [39].
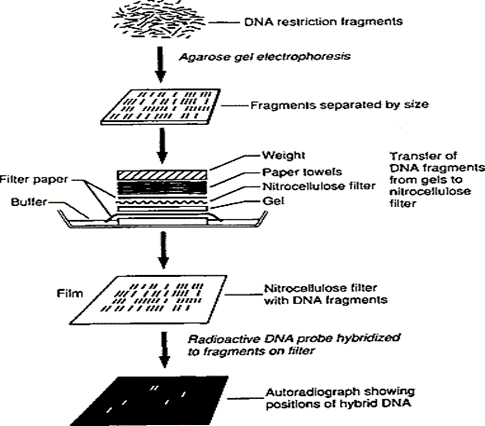
Figure 4. Procedure for Southern Blotting [40]
2.2.2. Northern Blotting. Northern blot is also a hybridization-based technique used for the detection of specific RNAs for several purposes. This technique is similar to southern blotting, except that the target sequence is an RNA molecule. Control over structure and function is achieved via northern blotting by identifying the level of expression of genes during differentiation and morphogenesis, as well as an abnormal condition. One of the major drawbacks is the degradation of mRNA during gel electrophoresis. Secondly, multiple probe detection makes the process less efficient [41].
2.2.3. In-situ Hybridization (ISH). In-situ hybridization (ISH) is an outstanding method for the identification and characterization of viral nucleic acids and other pathogens which are causative agents of different infectious diseases and cancer [42]. In-situ hybridization consists of four basic steps, namely (1) denaturation, (2) hybridization, (3) probe detection, and (4) analysis, as illustrated in Figure 5. ISH technique is used to regulate and detect the exact fixed location of different viruses, such as herpes simplex viruses, hepatitis viruses, human papillomaviruses, adenovirus, JC viruses, cytomegalovirus, and Epstein–Barr viruses [43]. The occurrence of cervical cancer is due to human papillomavirus (HPV). About 100 genomic types of HPV have been detected. Most types are not associated with the development of cervical cancer and hence are not oncogenic types. Therefore, those oncogenic types are divided into groups according to their potential to develop cancer, such as high-risk, moderate-risk, and low-risk HPVs. Currently, ISH is extensively used in cervical specimens for the differentiation and detection of HPV. There are different probes such as Dako Corporation supply probes for HPV ISH. These are named as biotinylated probes and high-risk groups [44].
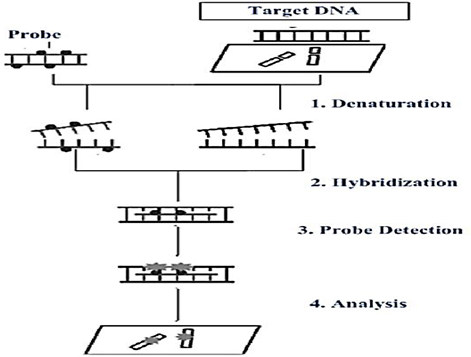
Figure 5. Basic Steps of In-Situ Hybridization [45]
2.2.4. Peptide Nucleic Acid-Fluorescence In-Situ Hybridization (PNA FISH). Fluorescence in situ hybridization (FISH) using peptide-nucleic acid (PNA) probes (PNA FISH) is a unique molecular technique. PNA probes have been used for the detection of infectious diseases quickly and with more accuracy. PNA probes are synthetic compounds with nucleotide bases the same as DNA that adhere to the backbone of peptides [45]. PNA with neutral backbone make PNA probes with great properties of hybridization, with a high level of specificity, rapid kinetics, strong affinity, and better hybridization ability for rRNA which is a highly structured target. PNA FISH probes have been made and checked for the identification of various pathogens, such as S. aureus, E. coli, E. faecalis, C. albicans, coagulase-negative staphylococci, Pseudomonas aeruginosa, C. dubliniensis and K. pneumoniae. Currently, FDA has approved some PNA probes for the pathogens listed above, such as S. aureus PNA FISH, C. albicans PNA FISH, and E. faecalis PNA FISH for diagnostic purposes. A recent evaluation suggests that PNA FISH is an accurate method with high sensitivity and specificity [46]. Mycobacterium bacteria can be identified from liquid culture with the help of using PNA FISH [47]. PNA FISH has been used also for the quick detection of species from positive cultures of blood and to evaluate bacterial diversity in environmental samples [48].
2.3. Sequence-based Technologies
Due to technological advancement in equipment and the lessening of price per reaction, DNA sequencing has been introduced in the laboratory. Up till now, sequencing has been utilized for the detection of microorganisms that have been hard to recognize by traditional methods. The target considered the utmost for that application is ribosomal DNA gene [49, 50].
2.3.1. Sanger Sequencing. Sanger sequencing was the key sequencing technique utilized during the period 1975-2005 and the golden standard for all sequencing technologies. The rRNA, particularly 16S and 23S rRNA, have been the most convenient phylogenetic markers for the diagnosis of diseases in Sanger sequencing [51]. The marker 16S rRNA is a widely used unit for microbiological species identification, and it is incredibly abundant in bacteria. The length of sequence 1 is about 1.5 kb [52] and it remains remarkably intact in terms of assembly and operation in a variety of flexible areas [53]. So, this sequence can be used for the identification of various genera and species of diverse pathogenic bacteria. At present, 16S rRNA bacterial gene analysis is mainly attained through sequencing the flexible regions of this gene [54]. The specific clones from microbes attained by the culture can be recognized through the Sanger method of sequencing, subsequently after amplification by 16S general primers. Bacterial, full-length, 16S rRNA gene could be sequenced via Sanger sequencing. Nonetheless, the disadvantage of this technique is that bacterial culture must be untainted; else, the bacterium could not be recognized. The positive rate of the clinical culture is not clearly known. Furthermore, microbes cannot be distinguished and the mix of the16S genes cannot be utilized to differentiate species. Moreover, it is a time-consuming technique. First-generation DNA sequencing is highly affected by all the above highlighted flaws [55]. An overview of Sanger sequencing is represented in Figure 6.
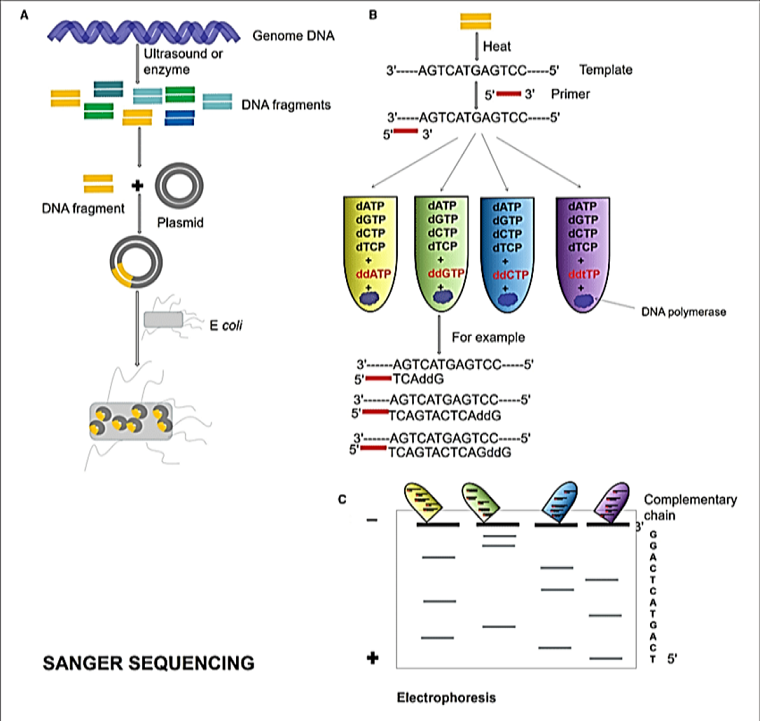
Figure 6. Overview of Sanger Sequencing [56]
2.3.2. MALDI-TOF MS. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is useful for the study of the entire bacterial protein profile. A few years ago, the sequencing of the nucleic acids of bacteria has been upgraded through MAL of the nucleic acid DI-TOF MS technique. Cleavage products have been studied by the MALDI-TOF MS the resultant mass patterns have been related to reference ranges for sequence purposes. Still, MALDITOF MS are probable to develop the process of choice for high-output testing. MALDITOF MS provides precise outcomes, is automatable, fast, and cost-effective [57, 58].
2.3.3. Multilocus Sequence Typing. Multilocus sequence typing (abbreviated as MLST) is an extension of MLEE. In MLST, alleles at all housekeeping loci are allocated directly through nucleotide sequencing. Interior -150 base pair fragments of the housekeeping genes are applied in MLST. Meanwhile, such fragments could be precisely sequenced on every strand with a single primers pair and, in many species, would offer adequate difference to identify numerous alleles in a population. Therefore, bacterial strains are defined by a string of integers consistent with alleles at all housekeeping loci. The use of nucleotide sequencing for the allocation of alleles at the housekeeping loci is of advantage [59]. Firstly, sequencing reveals all differences at a locus, resulting in more numerous alleles per loci as compared to those revealed by the MLEE. Therefore, MLST attains high levels of discrimination by using seven loci. Secondly, the uniqueness of alleles becomes definite through sequence data in contradiction of MLEE, where similar electrophoretic mobility might imitate identical/similar sequences of nucleotide, or totally dissimilar sequences that encode enzymes which travel on the starch gel at an equal rate. Thirdly, the electronic movability of the DNA sequences permits some testing centers to characterize bacterial isolates by presenting sequences of seven gene fragments through the Internet to the chief MLST website, which grips frequently increasing databases of the allelic profiles of the isolates of species. Finally, sequences of the seven loci from 100-1000 isolates of all species could be applied to address the features of their evolutionary biology and population [60].
2.3.4. Nanopore Technology. Nanopore technology is an electrical signal-based Seq-technology [61]. Protein-based nanopores (such as microscopic pores that basically form the channels on the membrane) are packed in the synthetic membrane and absorbed in the electrophysiological solution, which permits the ionic current to pass through them. The flow of ionic current is restricted when the molecules such as DNA/RNA pass it, producing a distinctive modification in the current signals. In nanopore technology, signal is evaluated in real-time format to determine the strand of DNA/RNA base sequence, which is transitory through the pore that permits the whole DNA/RNA sequence to be determined [62, 63]. This technology efficiently reports the flaws of the NGS in the aetiological diagnostic field, directly sequencing DNA/RNA that reads above 1 Mb in length [64]. Nanopore Seq-technology eradicates the exertion and the time required for reverse transcription. Since RNA reverse transcription into cDNA is not essential, the nanopore technology may execute RNA sequencing unswervingly [65].
2.3.5. Pyrosequencing. Pyrosequencing is a DNA sequencing method founded on the identification of the released pyrophosphate synthesis of DNA. The reactions are based on enzymes; visible light is produced which is proportionate to the number of the unified nucleotides. Enzymatic cascade is initiated with the polymerization reaction of nucleic acid (DNA/RNA) in which inorganic pyrophosphate remains unconfined due to the incorporation of nucleotide by the polymerase. Unconfined PPi is later converted to ATP through ATP sulfurylase, which delivers energy to the luciferase that oxidizes luciferin and also produces light. As more nucleotides come to light, then template sequence could be identified [66]. The complete reaction from polymerization to the detection of light occurs in 3–4 seconds at room temperature. Single PMOL of DNA in a pyrosequencing reaction produces 6 × 1011 molecules of ATP, which produce above 6 × 109 photons at 560 nm (wavelength). The light amount is simply detected through a photomultiplier tube, photodiode, and charge-coupled device camera. Two different strategies of pyrosequencing are now accessible, namely liquid-phase pyrosequencing [67] and solid-phase pyrosequencing [68]. Pyrosequencing has opened novel avenues for the accomplishment of sequence-based analysis of DNA. It is used for the analysis of SNPs (single nucleotide polymorphisms) and currently applied for the fast typing of a huge number of bacteria, viruses, and yeasts. Moreover, it remains the quickest technique for the sequencing of PCR products [69]. This technique is also used for the resequencing of suppressor gene (p53 tumor) where mutations are effectively determined and measured [70]. It has possible advantages of accuracy, parallel processing, flexibility, and automaticity.
2.4. Post-amplification Techniques
2.4.1. Array-based Techniques. Typical solid phase hybridization uses precise hybridization probes attached to the solid substance for the detection of target-labelled molecules in the solution. Primarily, assays for identification and characterization, certain probes were placed in microtiter plate wells and rendered immobile [71]. Secondarily, microarrays of the fixed manifold probes at definite locations on nitrocellulose/nylon membranes are advanced, with diverse probes applied as dot (in dot-blots)/lines (in line probe assay) [72, 73]. The diversity of numerous LIPA assays remains accessible for the detection of Enterococci that is vancomycin-resistant, the toxin of Clostridium difficile, MRSA, Neisseria meningitides, E. coli, and extended-spectrum and metallo-beta-lactamases. In comparison to microarrays, the DNA microarrays have reduced versions with spot sizes typically below 200-300 µm. These microarrays are typically restricted to less than a hundred probes. DNA microarrays differ from low-density arrays that carry 100-1000 probes, while high-density arrays comprise 10000-100000 probes. Various labelling and detection systems can be utilized in combination with array-based technologies, extending from the fluorescence-ended silver ppt to assessable enzyme-facilitated substrate alteration [74]. Peptide or protein arrays have been defined for the detection of the HCV/HIV and Mycobacterium tuberculosis [75, 76]. Microarrays antigen are used for the identification of the negative endocarditis blood culture [77]. DNA microarrays are applied for the diagnosis of bacterial meningitis and its general representation, as illustrated in Figure 7 [78].
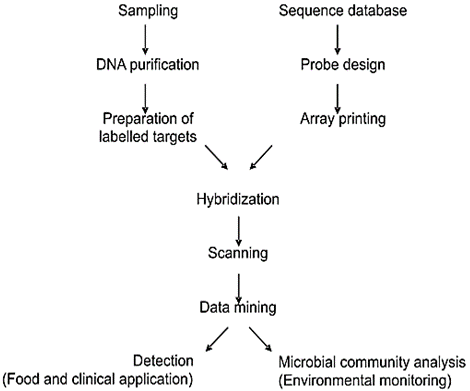
Figure 7. General Representation of the Microarray Experiment [8]
2.4.2. Loop-mediated Isothermal Amplification. It is a novel and strong amplification technique for nucleic acids and considered as a substitute for PCR. LAMP amplifies targeted nucleic acid with high specificity in isothermal conditions. In LAMP, there is no need for a thermal cycle for the changes made in temperature because it needs a specific temperature for DNA amplification [79]. Moreover, four primers comprising two internal and two external primers and Bst DNA polymerase are used to identify the six exclusive target sequences on the tested DNA. The two internal primers are the forward internal primer and the backward internal primer, while external primers are B3 and F3 [80]. Each FIP and BIP comprise two discrete sequences conferring to the sense or antisense strands of the targeted DNA. LAMP reaction begins with the help of one of the internal primers and the other is used for self-priming. This reaction is processed for 1 h in a water bath or heat block at 65°C. The amplification product is detected through SYBR Green 1 dye. The product yield has numerous inverted repeats of the targeted sequence that display a cauliflower-like assembly with many loops. It has 10 times better accuracy and sensitivity than traditional polymerase chain reactions [81]. Due to significant amplification efficacy, 1039 copies of the target part could be achieved within 1 hour of incubation. In LAMP, less inhibition reaction happens as compared to PCR in which molecular beacons act as detectors for the amplification product. A new process founded on MB-LAMP has been confirmed that offers direct detection of the LAMP product [82].
3. CONCLUSION
Studies about the history of evolving bacterial infections provide a new vision of the specific roles performed by various identification technologies. The old techniques are regarded as outdated due to the development of methods based on molecules. In this review, the use of direct hybridization, nucleic acid amplification, and variety of methods for postamplification analysis was made to detect microbial pathogens in human beings. PCR assists in identifying the taxonomical location of new organisms that still remain uncultured. LAMP is considered as a substitute for PCR and amplifies targeted nucleic acid with high specificity in isothermal conditions. Pyrosequencing technique is used for the resequencing of suppressor gene where mutations are effectively determined and measured. For the wide range and specific detection of pathogenic microbes, advancements in nucleic acid-based assay and sequencing have been introduced.
CONFLICT OF INTEREST
The author of the manuscript has no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
DATA AVALIABILITY STATEMENT
The data associated with this study will be provided by the corresponding author upon request.
References
- Reta DH, Tessema TS, Ashenef A, et al. Molecular and immunological diagnostic techniques of medical viruses. Int J Microbiol. 2020;2020:e8832728. https://doi.org /10.1155/2020/8832728
- Jung D, Liu L, He S. Application of in situ cultivation in marine microbial resource mining. Marine Life Science & Technology. 2020;3(2):148-161. https://doi.org/10.1007/s42995-020-00063-x
- Zhang J, Yang H, Li J, et al. Current Perspectives on Viable but Non-Culturable Foodborne Pathogenic Bacteria: A Review. Foods. 2023;12(6):1179-1179. https://doi.org/10.3390/foods12061179
- Umesha S, Manukumar HM. Advance molecular diagnostic techniques for the detection of food borne pathogens; Current applications and future challenges. Cri Rev Food Sci Nutr. 2018;58(1):84–104. https://doi.org /10.1080/10408398.2015.1126701
- Zhang J, Li C, Rahaman MM, et al. A comprehensive review of image analysis methods for microorganism counting: from classical image processing to deep learning approaches. Artificial Intelligence Review. 2021;55. https://doi.org/10.1007/s10462-021-10082-4
- Imran Ul Haq, Ijaz S, Iqrar Ahmad Khan. Phytomycology and Molecular Biology of Plant Pathogen Interactions. CRC Press; 2022.
- Liu Q, Jin X, Cheng J, Zhou H, Zhang Y, Dai Y. Advances in the application of molecular diagnostic techniques for the detection of infectious disease pathogens (Review). Molecular Medicine Reports. 2023;27(5). https://doi.org/10.3892/mmr.2023.12991
- Venbrux M, Crauwels S, Rediers H. Current and emerging trends in techniques for plant pathogen detection. Front Plant Sci. 2023;14:1120968-1120968. https://doi.org/10.3389/fpls.2023.1120968
- Aslam S, Tahir A, Aslam MF, Aslam MW, Shedayi AA, Sadia S. Recent advances in molecular techniques for the identification of phytopathogenic fungi-a mini review. J Plant Interact. 2017;12(1):493–504. https://doi.org/ 10.1080/17429145.2017.1397205
- Henson JM, French R. The polymerase chain reaction and plant disease diagnosis. Ann Rev Phytopathol. 1993;31(1):81–109.
- Bhat R, Browne G. Specific detection of Phytophthora cactorum in diseased strawberry plants using nested polymerase chain reaction. Plant Pathol. 2010;59(1):121–129. https://doi.org/10.1111/j.1365-3059.2009.02147.x
- Jabbar A, Zulfiqar F, Mahnoor M, Mushtaq N. Advances and perspectives in the application of CRISPR-Cas 9 in Livestocks. Mol Biotechnol. 2021; 63(1):757–767. https://doi.org/10.1007/s12033-021-00347-2
- Siqueira JF, Rocas NI. PCR methodology as a valuable tool for identification of endodontic pathogens. J Dent. 2003;31:333–339. https://doi.org/10.1016/S0300-5712(03)00051-4
- Rahman MT, Uddin MS, Sultana R, Moue A, Setu M. Polymerase chain reaction (PCR): a short review. Anwer Khan Modern Medical College J. 2013;4(1):30–36. https://doi.org/10.3329/akmmcj.v4i1.13682
- Trtkova J, Raclavsky V. Molecular-genetic approaches to identification and typing of pathogenic Candida yeasts. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2006;150(1):51–61.
- Tsai HL, Huang LC, Ann PJ, Liou RF. Detection of orchid Phytophthora disease by nested PCR. Bot Stud. 2006;47(4):379–387.
- Jaeger EE, Carroll NM, Choudhury S, et al. Rapid detection and identification of Candida, Aspergillus, and Fusarium species in ocular samples using nested PCR. J Clin Microbiol. 2000;38(8):2902–2908. https://doi. org/10.1128/jcm.38.8.2902-2908.2000
- Capote N, Aguado A, Pastrana AM, Sánchez-Torres P. Molecular tools for detection of plant pathogenic fungi and fungicide resistance. In: Cumagun CJ, ed. Plant Pathology. Intechopen; 2012;151–202.
- Eshaque B, Dixon B. Technology platforms for molecular diagnosis of cystic Õbrosis. Biotechnol Adv. 2006;24(1):86–93. https://doi.org/ 10.1016/j.biotechadv.2005.08.003
- De Clerck E, Van Mol K, Jannes G, Rossau R, De Vos P. Design of a 5¢ exonuclease-based real-time PCR assay for simultaneous detection of Bacillus licheniformis, members of the ‘ cereus group’ and B. fumarioli in gelatine. Lett Appl Microbiol. 2004;39(1):109–115. https://doi.org/ 10.1111/j.1472-765X.2004.01550.x
- Massart S, De Clercq D, Salmon M, Dickburt C, Jijakli MH. Development of real-time PCR using Minor Groove Binding probe to monitor the biological control agent Candida oleophila (strain O). J Microbiol Methods. 2005;60(1):73–82. https:// doi.org/10.1016/j.mimet.2004.08.012
- Bottger EC. Rapid determination of bacterial ribosomal RNA sequences by direct sequencing of enzymatically amplified DNA. FEMS Microbiol Lett. 1989;65(1-2):171–176. https://doi.org/ 10.1111/j.1574-6968.1989.tb03617.x
- Schabereiter-Gurtner C, Nehr M, Apfalter P, Makristathis A, Rotter ML, Hirschl AM. Evaluation of a protocol for molecular broad-range diagnosis of culture-negative bacterial infections in clinical routine diagnosis. J Appl Microbiol. 2008;104(4):1228–1237. https://doi.org/10.1111/j.1365-2672.2007.03648.x
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med. 1992;327(5):293–301. https://doi.org/1056/NEJM19920730327050
- Kornreich BG, Craven M, McDonough SP. Fluorescence in-situ hybridization for the identification of bacterial species in archival heart valve sections of canine bacterial endocarditis. J Comp Pathol. 2012;146(4):298–307. https://doi.org/ 10.1016/j.jcpa.2011.07.006
- Shivaprasad HL, Cadenas MB, Diab SS. Coxiella-like infection in psittacines and a toucan. Avian Dis. 2008;52(3):426–432. https://doi.org/ 10.1637/8192-120707-Reg
- Healy M, Huong J, Bittner T, Lising M, Frye S, Raza S. Microbial DNA typing by automated repetitive-sequence-based PCR. J Clin Microbiol. 2005;43(1):199–207. https://doi.org/10.1128/jcm.43.1.199-207.2005
- Sabat AJ, Budimir A, Nashev D, Sa-Leao R, Van Dijl J, Laurent F. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance. 2013;18(4):e20380. https://doi.org/10.2807/ese.18.04.20380-en
- Brossier F, Micaelo M, Luyt CE, Lu Q, Chastre J, Arbelot C. Could the DiversiLab(R) semi-automated repetitive-sequence-based PCR be an acceptable technique for typing isolates of Pseudomonas aeruginosa? An answer from our experience and a review of the literature. Can J Microbiol. 2015;61(12):903–912. https://doi.org/10.1139/cjm-2015-0372.
- Harrington SM, Stock F, Kominski A, Campbell J, Hormazabal J, Livio S. Genotypic analysis of invasive Streptococcus pneumoniae from Mali, Africa, by semiautomated repetitive-element PCR and pulsed-field gel electrophoresis. J Clin Microbiol. 2007;45(3):707–714. https://doi.org /10.1128/jcm.01871-06
- Sobral D, Mariani-Kurkdjian P, Bingen E, Vu-Thien H, Hormigos K, Lebeau B. A new highly discriminatory multiplex capillary-based MLVA assay as a tool for the epidemiological survey of Pseudomonas aeruginosa in cystic fibrosis patients. Eur J Clin Microbiol Infect Dis. 2012;31:2247–2256. https://doi.org/10.1007/s10096-012-1562-5
- Keim P, Price LB, Klevytska AM, Smith KL, Schupp JM, Okinaka R. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol. 2000;182(10):2928–2936. https://doi.org/10.1128/jb.182.10.2928-2936.2000
- Chen JW, Lau YY, Kishnan T, Chan KG, Chang CY. Recent advances in molecular diagnosis of Pseudomonas aeruginosa infection by State-of-the-art genotyping techniques. Front Microbiol. 2018;9:e378358. https://doi.org/10.3389/fmicb.2018.01104
- Ramamurthy M, Alexander M, Aaron S. Comparison of a conventional polymerase chain reaction for the detection of neurotropic viruses in cerebrospinal fluid samples. Ind J Med Microbiol. 2011;29(2):102–109. https://doi.org/10.4103/0255-0857.81777
- Souf S. Recent advances in diagnostic testing for viral infections. Biosci Horizons. 2016;9: https://doi.org/10.1093/biohorizons/hzw010
- Nakhaie M, Soleimanjahi H, Mollaie H, Arabzadeh S. Development of multiplex reverse transcription-polymerase chain reaction for simultaneous detection of influenza A, B and adenoviruses. Iran J Pathol. 2018;13(1):54–62.
- Shen M, Zhou Y, Ye J, et al. Recent advances and perspectives of nucleic acid detection for coronaviruses. J Pharm Anal. 2020;10(2):97–101. https://doi.org/10.1016/j.jpha.2020.02.010
- Boppana SB, Ross SA, Shimamura M, et al. Saliva polymerase-chain-reaction assay for Cytomegalovirus screening in newborns. New Eng J Med. 2011;364(22):2111–2118. https://doi.org/1056/NEJMoa1006561
- Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98(3):503–517. http://www.theses.ulaval.ca/2004/21483/21483014.png
- Bhagavan NV. Medical Biochemistry. Acadmic Press; 2002.
- Berg JM, Tymoczko JL, Stryer L. Biochemistry (Loose-Leaf). Macmillan; 2007.
- Wu TC, Lee WA, Pizzorno MC, et al. Localization of the human cytomegalovirus 2.7-kb major early ß-gene transcripts by RNA in situ hybridization in permissive and non-permissive infections. Am J Pathol. 1992;141(5):1247–1254.
- Li JJ, Huang YQ, Cockerell CJ. Localization of human herpes-like virus type 8 in vascular endothelial cells and perivascular spindle-shaped cells of Kaposi’s sarcoma lesions by in situ hybridization. Am J Pathol. 1996;148(6):1741–1748.
- Prange E, Trautmann JC, Kreipe H. Detection of Epstein-Barr virus in lymphoid tissues of patients with infectious mononucleosis by in situ hybridization. J Pathol. 1992;166(2):113–119. https://doi.org/ 10.1002/path.1711660206
- Ratan ZA, Zaman SB, Mehta V, Haidere MF. Application of fluorescence in-situ hybridization (FISH) technique for the detection of genetic aberration in medical science. Cureus. 2017;9(6):e1325. https://doi.org/10.7759/cureus.1325
- Wilson DA, Joyce MJ, Hall LS, et al. Multicenter Evaluation of a Candida albicans peptide nucleic acid fluorescent in situ hybridization probe for characterization of yeast isolates from blood cultures. J Clin Microbiol. 2005;43(6):2909–2912. https://doi.org /10.1128/jcm.43.6.2909-2912.2005
- Yeo SF, Wong B. Current status of nonculture methods for diagnosis of invasive fungal infections. Clin Microbiol Rev. 2002;35(3):465–484. https://doi.org/10.1128/cmr.15.3.465-484.2002
- Stender H. PNA FISH: an intelligent stain for rapid diagnosis of infectious diseases. Expert Rev Mol Diagn. 2003;3(5):649–655. https://doi.org/10. 1586/14737159.3.5.649
- Cai H, Archambault M, Prescott JF. 16S ribosomal RNA sequence-based identification of veterinary clinical bacteria. J Vet Diagn Invest. 2003;15(5):465–469. https://doi.org/ 10.1177/104063870301500511
- Woo PC, Ng KH, Lau SK. Usefulness of the MicroSeq 500 16S ribosomal DNA-based bacterial identification system for identification of clinically significant bacterial isolates with ambiguous biochemical profiles. J Clin Microbiol. 2003;41(5):1996–2001 https://doi.org/10.1128/ jcm.41.5.1996-2001.2003
- Ludwig W, Schleifer KH. Bacterial phylogeny based on 16S and 23S rRNA sequence analysis. FEMS Microbiol Rev. 1994;15(2-3):155–173. https://doi.org/10.1111/j.1574-6976.1994.tb00132.x
- Clarridge JE. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17(4):840–862. https://doi.org/10.1128 /cmr.17.4.840-862.2004
- Yarza P, Yilmaz P, Pruesse E. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–645. https:// doi.org/10.1038/nrmicro3330
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual‐index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–5120. https://doi.org/10.1128/AEM.01043-13
- Yu X, Jiang W, Shi Y, Ye H, Lin J. Applications of sequencing technology in clinical microbial infection. J Cell Mol Med. 2019;23(11):7134–7150. https://doi.org/10.1111/jcmm.14624
- Zang L, Chen F, Zeng Z, Xu M. Advances in Metagenomics and its application in environmental microbiology. Frontiers Microbiol. 2021;12:e766364. https://doi.org/ 10.3389/fmicb.2021.766364
- Von Wintzingerode F, Bocker S, Schlotelburg C. Base-specific fragmentation of amplified 16S rRNA genes analyzed by mass spectrometry: a tool for rapid bacterial identification. Proc Natl Acad Sci. 2002;99(10):7039–7044. https://doi. org/10.1073/pnas.102165899
- Lowe CA, Diggle MA, Clarke SC. A single nucleotide polymorphism identification assay for the genotypic characterization of Neisseria meningitidis using MALDI-TOF mass spectrometry. Br J Biomed Sci. 2004;61(1):8–10. https://doi.org/ 10.1080/09674845.2004.11732638
- Nakano K, Shiroma A, Shimojii M. Advantages of genome sequencing by long‐read sequencer using SMRT technology in medical area. Hum Cell. 2017;30:149–161. https://doi.org/ 10.1007/s13577-017-0168-8
- Spratt BG, Maden MC. Bacterial population genetics, evolution and epidemology. Philos Trans R Soc Lond B Biol Sci. 1999;354(1384):701–710. https://doi.org/10.1098/rstb.1999.0423
- Varongchayakul N, Song J, Meller A, Grinstaff MW. Single‐molecule protein sensing in a nanopore: a tutorial. Chem Soc Rev. 2018;47:8512–8524. https://doi.org/10.1039/ C8CS00106E
- Squires AH, Gilboa T, Torfstein C. Single‐molecule characterization of DNA‐protein interactions using nanopore biosensors. Methods Enzymol. 2017;582:353–385. https:// doi.org/10.1016/bs.mie.2016.08.010
- Tedersoo L, Tooming‐Klunderud A, Anslan S. PacBio metabarcoding of Fungi and other eukaryotes: errors, biases, and perspectives. New Phytol. 2018;217:1370–1385. https://doi.org/ 10.1111/nph.14776
- Van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The third revolution in sequencing technology. Trends Genet. 2018;34(9):666–681. https://doi.org /10.1016/j.tig.2018.05.008
- Marinov GK. On the design and prospects of direct RNA sequencing. Brief Funct Genom. 2017;16(6):326–335. https://doi.org/10.1093/ bfgp/elw043
- Benkovic SJ, Cameron CE. Kinetic analysis of nucleotide incorporation and misincorporation by Klenow fragment of Escherichia coli DNA polymerase I. Methods Enzymol. 1995;262:257–269. https://doi.org/ 10.1016/0076-6879(95)62022-2
- Ronaghi M, Pettersson B, Uhlen M, Nyren P. PCR-introduced loop structure as primer in DNA sequencing. 1998;25(8):876–884. https://doi.org/ 10.2144/98255rr02
- Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem.1996;242:84–89.
- Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2021;11:3–11. https://doi.org/10 .1006/abio.1996.0432
- Garcia AC, Ahamdian A, Gharizadeh B, Lundeberg J, Ronaghi M, Nyren P. Mutation detection by Pyrosequencing: Sequencing of exons 5 to 8 of the p53 tumor suppressor gene. 2000;253(2):249–257. https://doi.org/10.1016/S0378-1119(00)00257-2
- Van der Pol B, Quinn TC, Gaydos CA, et al. Multicenter evaluation of the AMPLICOR and automated COBAS AMPLICOR CT/NG tests for detection of Chlamydia trachomatis. J Clin Microbiol. 2000;38:1105–1112. https://doi.org/10.1128/jcm.38.3.1105-1112.2000
- Tortoli E, Mariottini A, Mazzarelli G. Evaluation of INNO-LiPA Mycobacteria v2: improved reverse hybridization multiple DNA probe assay for mycobacterial identification. J Clin Microbiol. 2003;41(9):4418–4420. https://doi.org/10.1128 /jcm.41.9.4418-4420.2003
- Jardi R, Rodriguez-Frias F, Tabernero D, et al. Use of the Novel INNO-LiPA Line Probe Assay for Detection of Hepatitis B Virus Variants That Confer Resistance to Entecavir Therapy. J Clin Microbiol. 2009;47(2):485–488. https://doi.org/10.1128/jcm.01678-08
- Martin DH, Cammarata C, Van Der Pol B, et al. Multicenter evaluation of AMPLICOR and automated COBAS AMPLICOR CT/NG tests for Neisseria gonorrhoeae. J Clin Microbiol. 2000;38(10):3544–3549.
- Burgess ST, Kenyon F, O’Looney N, et al. A multiplexed protein microarray for the simultaneous serodiagnosis of human immunodeficiency virus/hepatitis C virus infection and typing of whole blood. Anal Biochem. 2008;382(1):9–15. https://doi.org/10.1016/j.ab.2008.07.017
- Gaseitsiwe S, Valentini D, Mahdavifar S, et al. Pattern recognition in pulmonary tuberculosis defined by high content peptide microarray chip analysis representing 61 proteins from tuberculosis. PLoS ONE. 2008;3(12):e3840. https://doi.org /10.1371/journal.pone.0003840
- Gouriet F, Samson L, Delaage M, et al. Multiplexed whole bacterial antigen microarray, a new format for the automation of serodiagnosis: the culture-negative endocarditis paradigm. Clin Microbiol Infect. 2008; 14:1112–1118. https://doi.org/ 10.1111/j.1469-0691.2008.02094.x
- Ben RJ, Kung S, Chang FY, Lu JJ, Feng NH, Hsieh YD. Rapid diagnosis of bacterial meningitis using a microarray. J Formos Med Assoc. 2008;107(6):448–453. https://doi.org /10.1016/S0929-6646(08)60152-7
- Tsui CK, Woodhall J, Chen W, et al. Molecular techniques for pathogen identification and fungus detection in the environment. IMA Fungus. 2011;2(2):177–189. https://doi.org/10.5598/imafungus.2011.02.02.09
- Fakruddin M. Loop mediated isothermal amplification (LAMP)-an alternative to polymerase chain reaction (PCR). Bangladesh Res Pub J. 2011;5(4):425–439.
- Ren CH, Hu CQ, Luo P, Wang QB. Sensitive and rapid identification of Vibrio vulnicus by loop-mediated isothermal amplification. Microbiol Res. 2009;164(5):514–521. https://doi. org/10.1016/j.micres.2008.05.002
- Liu W, Huang S, Liu N, et al. Establishment of an accurate and fast detection method using molecular beacons in loop-mediated isothermal amplification assay. Sci Rep. 2017;7:e40125. https://doi.org/ 10.1038/srep40125
{kind=link}