Unveiling Cardiovascular Connections between Familial Hypercholesterolemia (FH) and Left Ventricular Hypertrophy (LVH)
Zoha Khan1*, Muhammad Suleman2, Atif Maqsood1, Bisma Bashir3, Muhammad Awais3, and Muhammad Shahbaz Aslam1
1School of Biochemistry and Biotechnology, 54590, University of the Punjab, Lahore Pakistan
2School of Biological Sciences, University of the Punjab, Lahore, Pakistan
3Faculty of Science and Technology, University of Central Punjab, Lahore, Pakistan
ABSTRACT
Left ventricular hypertrophy (LVH), a complex cardiac condition characterized by the enlargement and thickening of the left ventricle, is primarily associated with hypertension and valvular heart disease. Recent studies have identified familial hypercholesterolemia (FH) as a secondary cause of LVH. It is characterized by high low-density lipoprotein cholesterol (LDL-C) in blood. FH is an inherited disorder which involves genetic variations associated with abnormal metabolism of LDL-C. This review article aims to provide a comprehensive overview of the relationship between FH and LVH. It summarizes the current understanding of the pathophysiological mechanisms underlying this association and discusses its implications for clinical practice. Elevated LDL-C levels in FH patients lead to accelerated atherosclerosis and an increased risk of premature cardiovascular events. Animal models and clinical observations provide insights into the mechanistic links between elevated LDL-C levels, oxidative stress, inflammation, and LVH development. Early diagnosis of FH would certainly play a critical role in preventing or delaying the development of LVH and subsequent cardiovascular complications. Preemptive measures emphasize the identification of at risk individuals, in-depth clinical evaluations, and implementation of effective treatments including lifestyle modifications, statins, and adjunctive therapies, such as PCSK9 inhibitors or lipoprotein apheresis. By increasing the awareness of FH as a secondary cause of LVH, healthcare professionals can improve early detection and implement appropriate management strategies to mitigate the cardiovascular burden associated with this inherited disorder.
Highlights
• Left ventricular hypertrophy (LVH), often tied to hypertension and valvular heart disease, is also associated with familial hypercholesterolemia (FH), an inherited disorder marked by high LDL-C levels due to gene mutations in LDL-C metabolism proteins.
• Elevated LDL-C in FH leads to accelerated atherosclerosis, linking oxidative stress, inflammation, and LVH development. Animal models and clinical observations provide insight into the intricate connections between these factors.
• Early identification and management of FH are vital to prevent LVH and other cardiovascular complications. Guidelines stress risk assessment, thorough evaluation, and aggressive lipid-lowering treatments including lifestyle changes, statins, and advanced therapies, as well as enhanced clinical practice awareness.
1. INTRODUCTION
Familial hypercholesterolemia (FH) is a genetically inherited condition that affects the metabolization of lipoproteins, leading to decreased LDL levels in the bloodstream. LDLR gene mutations are standard but apolipoprotein B and PCSK9 can produce similar results [1]. A 2019 study revealed that 9% of the world's 195 countries reported FH in their population. This leaves 178 nations with an unknown prevalence of this medical condition. Over time, high LDL levels can cause cholesterol to build up in vessels, eventually narrowing them [2].
Atherosclerosis causes the blood vessels in the kidneys to constrict, leading to a reduction in blood flow. This condition is known as renal artery stenosis (RAS). It is caused by vasoconstriction which signals increased renin production, activating the renin-angiotensin-aldosterone system and resulting in hypertension. This makes it difficult for the heart to meet the body's oxygen demand due to the increased afterload, causing hypertrophy of heart muscles. The prevalence of left ventricular hypertrophy (LVH) in hypertensive patients is between 23-48% [3].
1.1. Familial Hypercholesterolemia (FH)
It is a genetic disorder that puts the patients at risk of high LDL levels in blood, as well as the narrowing and hardening of the arterioles and the development of cholesterol plaque on the walls of arteries and underneath the skin (which is known as xanthoma). Moreover, FH results in the change of blood color from purple-red to strawberry-pink [4]. Its worldwide prevalence is 7 in 100 people with ischemic heart disease (IHD). According to a meta-analysis of 11 million subjects, it is 20-fold higher among those with premature IHD and 23-fold higher among those with severe hyperlipidemia 1. FH affects about 1 in 200 to 250 people in the US, according to a European report based on the Dutch lipid clinic criteria, which is available in the National Health and Nutrition Examination Survey (NHANES). The population prevalence of FH is about 1:1,000,000 in the case of homozygous and 1:500 in the case of heterozygous FH [5].
Research indicates that the LDLR gene codes for the low-density lipoprotein receptor - a protein that controls many low-density lipoproteins in the blood. Mutations in this gene can reduce its effectiveness, which causes FH [6]. LDLR gene comprises exons of the size 45 kb and encodes a glycoprotein made up of 839 amino acids, which is essential in cholesterol homeostasis. As reported, the loss of function mutations in the LDLR gene reduces LDL receptors' ability to uptake LDL cholesterol (LDL-C) from the blood [7]. Furthermore, mutations in the LDLR gene are classified based on their effect on receptor protein. LDL Class I mutations directly affect the synthesis of LDLR in the endoplasmic reticulum, whereas Class II mutations prevent their transport to the Golgi apparatus. Class III mutations avert LDL-C binding with receptors, while LDL Class IV mutations impede the internalization of the receptor-ligand complex. LDL Class V mutations lead to the production of receptors that cannot recycle properly [8].
To find out the gene mutations that cause FH and to determine their types and frequency, researchers conducted a study with FH patients from north-western Greece. Notably, 1775 G>A in exon 12 is the most frequent mutation (34%) in Greek [9], Italian, and German populations [10]. Moreover, 1646 G>A in exon 11 is the second most likely (22%), while 858 C>A is the third most common mutation in exon 6 of the LDLR gene (21.3%) in the north-western Greek population. Other LDLR gene mutations in the above populations include 81T>G (4.7%) in exon 2, 517 T>G (3.6%) in exon 4, 761 A>C in exon 5, and 1352 T>C,1195 G>A in exon 9 (0.6%) [11]. Furthermore, pathogenic variations in apolipoprotein (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes were also reported, which were found to be responsible for FH. APOB gene, located on chromosome 2 with 29 exons, produces two isoforms namely APOB-100 and APOB-48. Mutations in APOB-100, being similar to FH in phenotypes, are termed as familial defective APOB-100 (FDB) [12]. Notably, 5% of clinically diagnosed cases of FH are attributable to FDB, with a prevalence of 1 in 500 in population studies [13]. The most common mutation in exon 26 of the APOB gene is p. R3527Q which accounts for 6-10% of FH cases in the European population [14]. Other APOB gene mutations, including p.Arg50Trp, p.Arg116Thr, and p.Gln4494del were also found to cause FH [15].
Additionally, the PCSK9 gene (located on chromosome 1 with 12 exons) produces an enzyme called proprotein convertase subtilisin/kexin type 9 (PCSK9). This gene is a part of the proprotein convertase family. Mutations that increase the function (GOF) of the PSCK9 gene can lead to lysosomal degradation of LDLR, as it binds to LDLR protein and stops it from being transported back to the cell surface. p.Ser127Arg and p.Asp374Tyr are the respective GOF mutations in PCSK9. These were found in 3 Norwegian and 3 English families [16] and account for 1-3% of clinically diagnosed FH. As far as the distribution of FH-related variances is concerned, out of 119 distinct variants 84 (70.58%) were in the LDLR gene with 48.74% non-synonymous mutations, 6.72% frame-shift mutations, 4.20% LDLR-splicing, and 10.92% stop gain mutations. Whereas, 26.05% mutations were in APOB and 3.36% in PCSK9 gene [17].
Research showed that FH patients from multi-generational families are more likely to face an increased risk of death due to gene-environment interactions based on a sedentary lifestyle and changes in dietary composition [18]. In addition to genetic differences, people with FH may have variations in their DNA methylation, which could mess with genes that control lipid levels. This can cause changes to the amount of lipids in the bloodstream of those with heterozygous FH mutations [19]. FH is rarely diagnosed in time, leading to a high cardiovascular disease burden. This review arose from the need to address gaps in the existing knowledge about the link between familial FH and LVH.
1.2. Etiology
Higher LDL levels than usual are due to mutations in LDLR, APOB, and PCSK9 genes [20]. These mutations make it harder for the body to clear cholesterol from the blood, leading to reduced internalization of LDL, reduction in LDL binding, lack of expression of receptors, and the failure of LDL to reach the plasma membrane. The uptake and removal of LDL from plasma is carried out by LDL receptors present on the cell membrane of hepatocytes. APOB, an LDL-bound protein, binds to LDL receptors and PCSK9 degrades LDL receptors. The LDL-C/APOB complex undergoes endocytosis by binding to the clathrin/LDLRAP1 pits. Then, LDL gets dissociated and degraded in the lysosome [21].
High cholesterol levels affect the systolic function of the heart through a gradual accumulation of cardiac lipids, leading to oxidative strands and mitochondrial dysfunction. FH decreases coronary blood flow by inducing capillary endothelial cell apoptosis, ultimately leading to impaired left ventricle function [22].
1.3. FH and Atherosclerosis
The elevated free cholesterol levels in FH alter the membrane function and enhance the production of reactive oxygen species (ROS) by modifying the caveolin-associated pathway [23]. Caveolin, an essential protein component of lipid rafts called caveolae, acts as a scaffolding protein by organizing the lipids within rafts. In FH, caveolin binds with endothelial nitric oxide synthase (eNOS) to cause eNOS inactivation, thus affecting the production of ROS. Consequently, nitric oxide (NO) bioavailability decreases due to the upregulation of caveolin abundance. NO takes part in the normal vascular endothelium function by inhibiting platelet aggregation, cellular adhesion, and proliferation of vascular smooth muscles. The decreased bioavailability of NO and an imbalance in ROS generation alters endothelial cells' anti-inflammatory properties by causing oxidative stress (Figure 1) and increasing vascular permeability [24].
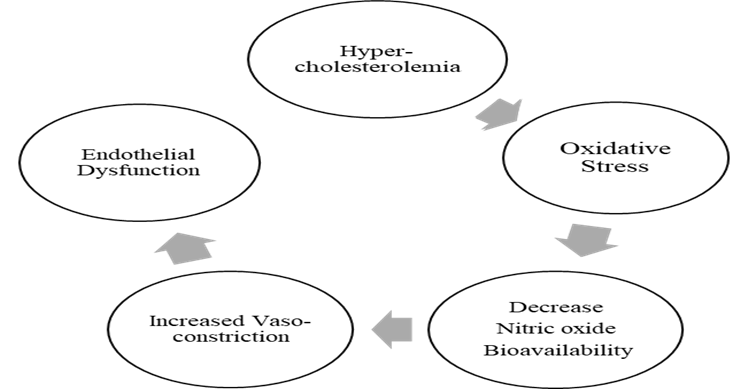
Figure 1. Hypercholesterolemia-Induced Endothelial Dysfunction
Endothelial dysfunction and vasoconstriction can happen due to elevated ROS production caused by increased caveolin. This also leads to increased levels of oxidized LDL, further reducing NO's bioavailability. The ROS released from dysfunctional endothelial cells activates the receptors for WBCs, after which leukocytes adhere to the cell wall, residing within the intimal space [25].
The macrophages then begin to accumulate oxidized LDL, which transforms into foam cells between the basal lamina and the smooth muscle layer, enhancing the production of oxidative stress markers and leading to vascular dysfunction [26]. The chemotactic activity of monocyte chemotactic protein-1 (MCP-1) and interleukin-8 (IL-8) for monocyte recruitment is important in FH patients, leading to wall remodeling. Foam cell formation and lipoprotein oxidation due to decreased NO bioavailability are the precursors of atherosclerotic plaque [27].
The deposited LDL in tunica media gets oxidized due to the release of ROS from dysfunctional endothelial cells, which further activates the receptors for WBCs. The adhesion of WBCs to receptors allows monocytes to enter tunica intima, become macrophages, and transform into foam cells by taking up oxidized LDL [28], thus forming fatty streaks (a yellowish lesion). The foam cells in the arteries’ mid-layer come from Smooth Muscle Cells (SMCs). They migrate to the innermost layer and increase collagen production, causing the hardening of atherosclerotic plaques. As part of this process, foam cells die and release fats, which can lead to blockages in the arteries. If they rupture, blood flow gets restricted and clots are formed. This is known as thrombosis [29] (shown in Figure 2). A brief description of vascular modifications during atherosclerosis is given in Table 1.
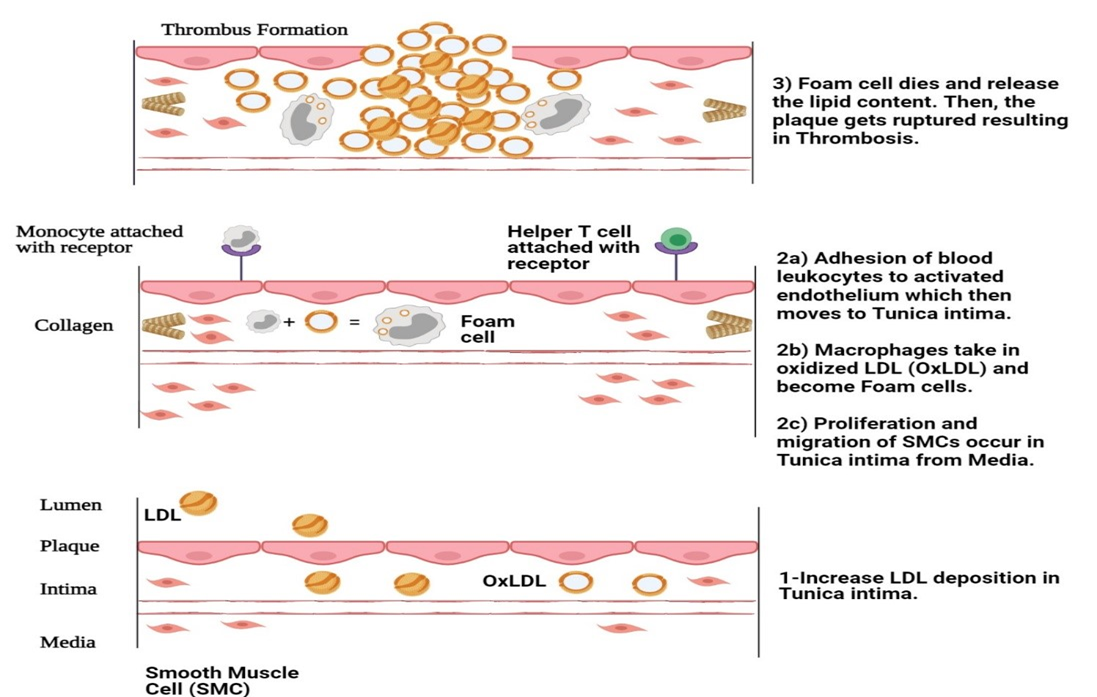
Figure 2. Thrombus and Foam Cell Formation in Vessels
Excess cholesterol gets deposited in tunica media. 2) Monocytes and leukocyte recruitment begins at the site of inflammation where monocytes with oxidized LDL form foam cells along with the proliferation of SMCs. 3) This results in a fibrous cap and thrombus formation upon plaque rupture. The above diagram was adapted from [30].
1.4. Atherosclerosis Prognosis to Hypertension
Atherosclerosis causes renal artery stenosis (RAS) in 90% of cases [31]. Valvular and supra-valvular aortic stenosis is observed in patients with homozygous FH (HoFH), along with the calcification and thickening of cusps. This narrows down the orifice of aortic valves, reducing the diameter of valsalva and post-stenotic dilatation of the ascending aorta [32]. Chronic ischemia develops due to obstruction in blood flow to the kidneys which results in decreased perfusion, thus activating the renin-angiotensin-aldosterone system which leads to renin production and maintains homeostasis. Renin is essential in producing ANG I & II, two powerful vasoconstrictors. ANG I is formed as a reaction to renin and ACE further generates ANG II from it within the lungs. Renin overproduction by juxtaglomerular cells in ischemic conditions increases the glomerular filtration rate (GFR) [33]. ANG II is responsible for vasoconstriction, thus raising blood pressure and stimulating the release of aldosterone from the adrenal gland. This causes salt and water retention and leads to the thickening of the vascular wall and fibrosis by increasing the synthesis of collagen type I and III [34]. Hypertension developed due to renal artery stenosis is termed as renovascular hypertension. Patients with progressive renal artery stenosis eventually develop end-stage renal disease (ESRD) [35]. The thickness of intima-media in coronary arteries increases the risk of myocardial infarction and cerebrovascular diseases. The aortic valve dysfunction in patients with homozygous FH suggests that the increased cholesterol levels affect coronary arteries as well as the aortic valve, in response to which LVH develops [36].
Table 1. Vascular Modifications in Atherosclerotic Disease
|
Vascular Modifications |
Characteristics |
References |
|
Pathologic intimal thickening |
Layers of smooth muscle cells and extracellular matrix aggregate near the lumen. |
[37] |
|
Foam cell formation (Fatty streak) |
Macrophages engulf oxidized LDL and are then termed as foam cells. Thus, abundant macrophage foam cells along with SMCs result in fatty streaks. |
[38] |
|
Fibrous cap atheroma |
Layers of fibrous connective tissues prone to rupture, resulting in thrombosis. |
[39] |
|
Atheromatous plaques |
A collection of macrophages, including cholesterol, which are highly unstable. |
[40] |
|
Ruptured plaque |
The disruption of luminal thrombus and fibrous caps. |
[41] |
1.5. Hypertension Leads to LVH
LVH is caused by high blood pressure and can also be genetically inherited. It increases muscle mass in the left ventricle and involves a considerable growth of sarcomeres within each myocardial cell. Two-dimensional echocardiography is commonly used to measure left ventricular accurately (LV mass). Generally, the extent of LVH is determined using wall thickness and cavity dimensions. Even if a patient's absolute heart mass hasn't increased in remodeling, changes such as a thicker wall or a larger wall-cavity diameter ratio can still pose potential risks. Those with increased mass and relative wall thickness have amplified danger levels [42]. The myocardium comprises three distinct compartments, namely the muscular, interstitial, and vascular compartments. The muscular compartment consists mainly of myocytes, while the interstitial compartment is formed by fibroblasts and collagen. On the other hand, the vascular compartment consists of smooth muscle cells and endothelial cells. Hypertension (HTN) can cause an increase in LV wall thickness. This triggers an augmented afterload on the myocardium, prompting the growth of myocytes, collagen formation, and fibroblasts, as well as the remodeling of cardiac tissue with an elevated level of fibrous tissue. These alterations can decrease LV compliance, resulting in impaired diastolic function. Moreover, the increased myocardial mass and fibrosis reduce coronary flow through the vessels.
Studies showed that people with HTN present an increased resistance of their arteries, which leads to arterial vessel enlargement and stiffness. This, in turn, is associated with an escalation of systolic blood pressure and eventually causes LVH. The latter’s development can significantly affect ventricular volume, wall thickness, and wall mass [43]. Overwhelming pressure results in an increase in myofibrils, which thickens the walls of the ventricle. This phenomenon, paired with the fact that end-diastolic volume remains unchanged, causes an increase in the ventricular weight-to-volume ratio. Secondly, in the case of increasing volume, the ventricular wall thickness remains unchanged or increases slightly. In contrast, ventricular weight and volume increase due to the replication of myofibrils, resulting in eccentric ventricular hypertrophy. Thirdly, LVH in obstructive cardiomyopathy leads to an increase in ventricular weight concerning volume due to extreme upsurge in wall thickness and muscle mass [44].
1.6. Pathophysiology of the Disease
In HTN, an increase in the amount of ANG II causes vasoconstriction, thus increasing the venous return to the heart and the central venous pressure. ANG II is also responsible for myocardial fibrosis and induces fibroblast proliferation, along with the stimulation of aldosterone that leads to collagen accumulation [45]. Arterial stiffness occurs due to increased collagen production and peripheral resistance. It may also be caused by elevation in sodium levels that directly affects vascular endothelium and the release of NO in patients with renal dysfunction [46].
In patients with elevated cholesterol levels, HTN and mechanical stress increase ROS production and alter the function of potassium channels in myocardium, where simple activation of these channels would inhibit the 70-KDa S6 kinase and trigger the protein synthesis in myocardial remodeling, thus reducing cardiac hypertrophy [47]. HTN promotes ischemic injury to the heart because of high oxygen demand and the Vessel’s tolerance decreases due to the increased pressure. Initially, cardiac output is maintained instead of increased LV wall thickness but it worsens when the condition prevails. Increased workload on the heart in prolonged hypertension and aortic stenosis leads to an abnormal increase in myocardial mass, resulting in concentric LVH [48].
1.7. Endothelial Dysfunction and Disease Association: Implications for Vascular Health
Endothelium regulates vascular wall homeostasis by maintaining a relaxed vascular tone and reduces oxidative stress by releasing mediators such as NO. Endothelium-dependent vasodilation gets impaired in hypercholesterolemia due to reduced bioavailability of NO and high cholesterol levels in the blood, leading to atherosclerosis [49].
Prolonged exposure to high cholesterol levels causes the formation of fatty streaks in arteries. Atherogenesis, initiated by oxidative stress and endothelial injury, creates an imbalance between vasodilation and vasoconstriction, hence stimulating RAAS by promoting cardiac fibrosis [50]. Fibrosis leads to the progressive weakening of contractility and stiffening of the myocardial wall. In patients with hypertensive heart disease, fibrosis facilitates the change from LVH to heart failure [51] (Figure 3).
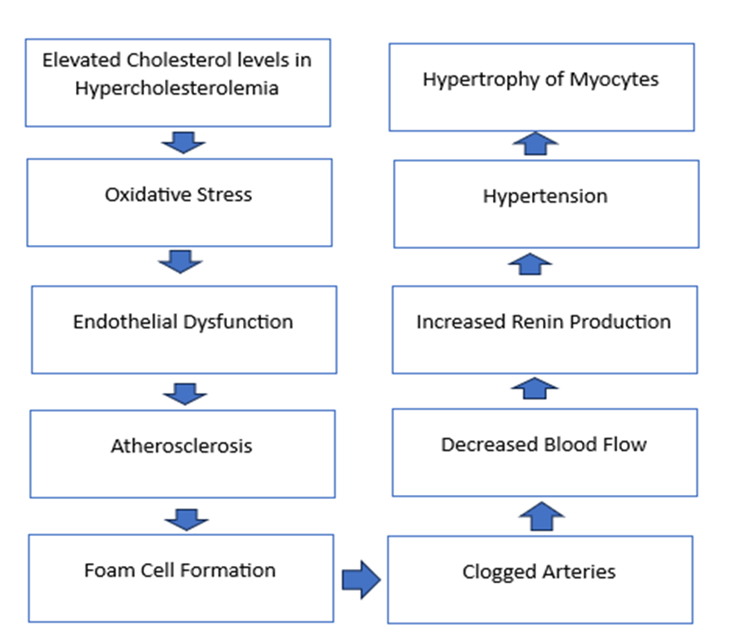
Figure 3. Disease Associations
Elevated cholesterol levels in hypercholesterolemia cause oxidative stress due to an imbalance in the generation of ROS, thus reducing NO bioavailability, leading to endothelial dysfunction predisposing to atherosclerosis. Macrophages engulf excess LDL and transform into foam cells, which clog up the arteries, causing aortic valve stenosis. Decreased blood flow signals increased renin production, ultimately resulting in HTN. Thus, hypertrophy of myocytes leads to LVH due to increased workload on the heart.
1.8. Treatments
The first line of defense against high cholesterol and associated cardiovascular diseases is a healthy lifestyle which includes daily exercise, cessation of tobacco products and alcohol consumption, reduced intake of high saturated fats (<7% of total energy intake), and the use of dietary cholesterol (200mg/d) [52] as diet treatment. This adds incremental health benefits to the pharmacological treatment.
Using plant stanols, sterol ester (2 g/d), and soluble fibers (10-20 g/d), such as peas, apples, oats, beans, and citrus fruits helps to maintain a healthy body weight and normal blood pressure [53]. In other treatments used to control the elevated levels of LDL in FH patients, statins are the drug of choice for HeFH treatment as they block the enzyme HMG-CoA reductase, a main enzyme for the synthesis of cholesterol in the liver, HMG-CoA reductase. Statins induce liver toxicity, thus damaging liver cells while increasing aspartate aminotransferase, alanine aminotransferase, and myalgia [54]. On the contrary, statins are not that much involved in HoFH, so combination therapies are preferred to enhance their efficacy [55].
Statins can be used in combination with ezetimibe, a cholesterol absorption inhibitor that blocks dietary cholesterol absorption and transfers intestinal cholesterol to the liver [56]. Despite this combination therapy, LDL-lowering drugs are required further in some cases, such as bile acid sequestrants that reduce the enterohepatic circulation of bile acids by binding with the cholesterol in the gut and enhancing its conversion to bile acids, ultimately increasing the removal of LDL from circulation. After statins and cholesterol absorption inhibitors, bile acid sequestrants are considered as the second-line therapy. Niacin is the next drug of choice if those mentioned earlier remain ineffective. It upturns HDL-C by 15-30% and lowers LDL-C and triglycerides by 20% and 35%, respectively [57].
Newly approved drugs by FDA for FH therapy include mipomersen, lomitapide, and PCSK9 inhibitors. Mipomersen (an APOB synthesis inhibitor) targets mRNA molecules encoding APOB-100 produced by the liver. It is being tested in populations with HoFH, HeFH, with coronary artery disease (CAD), people with severe hypercholesterolemia, and those at high risk of CAD with FH. It has shown significant reductions in LDL levels both on administration with other statins or as a single drug [58].
The microsomal triglyceride transfer protein (MTTP) is involved in lipid metabolism, VLDL, and the formation of chylomicrons. It is required for the secretion of VLDL and chylomicrons by the liver and intestine, respectively. It is involved in the loading of triglycerides on APOB-100. Lomitapide binds MTTP and inhibits it, thus reducing plasma levels of APOB-containing lipoproteins. Both mipomersen and lomitapide are hepatotoxic and can only be prescribed by certified healthcare workers.
PCSK9 inhibitor (PCSK9i), a serine protease inhibitor, leads to high levels of LDL-C by degrading LDL receptors and decreasing circulating LDL-C levels [59]. Secreted PCSK9 binds to LDLR on the surface of hepatocytes, thus reducing their number on the cell surface by enhancing the internalization and degradation of LDLR in lysosomes. So, secreted PCSK9 is inhibited, increasing the number of LDLRs on the cell surface and increasing LDL-C uptake, thus lowering LDL-C [60]. For treating HTN-induced LVH, combinations of antihypertensive drugs are used. PCSK9 monoclonal antibodies play a pivotal role in regulating cholesterol homeostasis, as they bind to LDL-R on the surface of hepatocytes and thus, interfere with LDL clearance in circulation [61]. ACE inhibitors seem to be effective in LVH patients as they improve the release of NO, thereby decreasing the oxygen consumption by myocardial cells via NO inhibition of mitochondrial respiration [62]. Amlodipine, a calcium channel blocker, facilitates the regression of LVH and blood pressure regulation when combined with ARBs and ACE inhibitors [63]. Diuretics, such as spironolactone, facilitate the regression of LVH and also reduce fibrosis in it. Thus, diuretics and CCBs combine with ACE inhibitors or ARBs to improve the coronary blood flow [64].
4. CONCLUSION
FH is the most common genetic disorder with life-threatening cardiovascular consequences. Early initiation of lipid-lowering therapy and lifestyle measures reduce the clinical outcome. There is an increased risk of developing RAS due to endothelial dysfunction that eventually leads to LVH in patients with hypercholesterolemia-induced HTN. For plaque stabilization in these patients, treatment should be started with statins and RAAS inhibitors to control HTN. Statins contribute to a significant decrease in blood pressure, whereas antihypertensive drugs play their role in reducing the atherogenicity of FH. Further, if treatment goals are unmet, then approved drugs (lomitapide, mipomersen, and PCSK9 inhibitors) by FDA should be considered. When the single-drug therapy fails, two-drug combinations of CCBs, thiazides, and other diuretics for more effective treatment regimens should be used. Further investigation is essential to comprehend the exact mechanism of these combination therapies and devise the best treatment.
CONFLICT OF INTEREST
The authors of the manuscript have no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
DATA AVALIABILITY STATEMENT
Data availability is not applicable as no new data was created.
REFERENCES
- Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide prevalence of familial hypercholesterolemia: meta-analyses of 11 million subjects. J Am Coll Cardiol. 2020;75(20):2553–2566. https://doi.org/10.1016/j.jacc.2020.03.057
- Miname MH, Santos RD. Reducing cardiovascular risk in patients with familial hypercholesterolemia: risk prediction and lipid management. Prog Cardiovasc Dis. 2019;62(5):414–422. https://doi.org/10.1016/j.pcad.2019.10.003
- Colbert GB, Szerlip HM. Cardiovascular Impact of Atherosclerotic Renovascular Disease. In: Rangaswami J, Lerma E, McCullough P. eds. Kidney Disease in the Cardiac Catheterization Laboratory. Springer; 2020:69–81. https://doi.org/10.1007/978-3-030-45414-2_4
- Kulkarni JD, Bhatia P, Pai SA. Strawberry pink blood. Indian J Hematol Blood Transfus. 2016;32(4):512–513. https://doi.org/10.1007/s12288-016-0695-6
- Harada-Shiba M, Arai H, Ishigaki Y, et al. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017. J Atheroscler Thromb. 2018;25(8):751–770. https://doi.org/10.5551/jat.CR003
- Mabuchi H. Half a century tales of Familial Hypercholesterolemia (FH) in Japan. J Atheroscler Thromb. 2017;24(3):189–207. https://doi.org/10.5551/jat.RV16008
- Durst R, Ibe UK, Shpitzen S, et al. Molecular genetics of familial hypercholesterolemia in Israel–revisited. Atherosclerosis. 2017;257:55–63. https://doi.org/10.1016/j.atherosclerosis.2016.12.021
- Tahmasebi-Birgani M, Zeydooni M, Abolfathi S, et al. Mutation screening of PCSK9 exons in Iranian Arab patients suffering from familial Hypercholesterolemia. SSRN Elect J. 2023:e4333995. http://dx.doi.org/ 10.2139/ssrn.4333995
- Guo X, Gao M, Wang Y, et al. LDL receptor gene-ablated hamsters: a rodent model of familial hypercholesterolemia with dominant inheritance and diet-induced coronary atherosclerosis. eBioMedicine. 2018;27:214–224. https://doi.org/10.1016/j.ebiom.2017.12.013
- Moradi A, Maleki M, Ghaemmaghami Z, et al. Mutational spectrum of LDLR and PCSK9 genes identified in Iranian patients with premature coronary artery disease and familial hypercholesterolemia. Front Genet. 2021;12:e625959. https://doi.org/10.3389/fgene.2021.625959
- Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24(1):133–170. https://doi.org/10.1146/annurev.ge.24.120190.001025
- Bea AM, Lamiquiz-Moneo I, Marco-Benedí V, et al. Lipid-lowering response in subjects with the p.(Leu167del) mutation in the APOE gene. Atherosclerosis. 2019;282:143–147. https://doi.org/10.1016/j.atherosclerosis.2019.01.024
- Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111(12):1795–1803. https://doi. org/10.1172/JCI18925
- Alves AC, Chora JR, Bourbon M. Genomics of familial hypercholesterolaemia. Curr Opin Lipidol. 2019;30(2):148–150. https:// doi.org10.1097/MOL.0000000000000584
- Blanco-Vaca F, Martin-Campos JM, Beteta-Vicente Á, et al. Molecular analysis of APOB, SAR1B, ANGPTL3, and MTTP in patients with primary hypocholesterolemia in a clinical laboratory setting: Evidence supporting polygenicity in mutation-negative patients. Atherosclerosis. 2019;283:52–60. https://doi.org/10.1016/j.atherosclerosis.2019.01.036
- Los B, Ferreira GM, Borges JB, et al. Effects of PCSK9 missense variants on molecular conformation and biological activity in transfected HEK293FT cells. Gene. 2023;851:e146979. https://doi.org/10.1016/j.gene.2022.146979
- Sun D, Zhou BY, Li S, et al. Genetic basis of index patients with familial hypercholesterolemia in Chinese population: mutation spectrum and genotype-phenotype correlation. Lipids Health Dis. 2018;17(1):e252. https://doi.org/10.1186/s12944-018-0900-8
- Truong TH, Do DL, Kim NT, Nguyen MN, Le TT, Le HA. Genetics, screening, and treatment of familial hypercholesterolemia: Experience gained from the implementation of the Vietnam familial hypercholesterolemia registry. Front Genet. 2020;11:e914. https://doi.org/10.3389/fgene.2020.00914
- Reeskamp LF, Venema A, Pereira JPB, et al. Differential DNA methylation in familial hypercholesterolemia. eBioMedicine. 2020;61:e103079. https://doi.org/10.1016/j.ebiom.2020.103079
- Khan MR, Batool M, Amir RM, et al. Ameliorating effects of okra (Abelmoschus esculentus) seed oil on hypercholesterolemia. Food Sci Technol. 2020;41(1):113–119. https://doi.org/10.1590/fst.38919
- Katzmann JL, Gouni-Berthold I, Laufs U. PCSK9 inhibition: insights from clinical trials and future prospects. Front Physiol. 2020;11:e595819. https://doi.org/10.3389/fphys.2020.595819
- Yao YS, Li T Di, Zeng ZH. Mechanisms underlying direct actions of hyperlipidemia on myocardium: an updated review. Lipids Health Dis. 2020;19(1):e23. https://doi.org/ 10.1186/s12944-019-1171-8
- Zhang Z, Wu H, Wang T, Liu Y, Meng C. Mechanisms of myocardial damage due to hyperlipidemia: a review of recent studies. Med Sci Monit. 2022;28:e937051. https://doi.org/10.12659%2FMSM.937051
- Peluso I, Urban L, Ioannone F, Serafini M. Oxidative stress in atherosclerosis development: the central role of LDL and Oxidative Burst. Endocr Metab Immune Disord Drug Targets. 2012;12(4):351–360. https://doi.org/10.2174/187153012803832602
- Tousoulis D, Kampoli A-M, Tentolouris Nikolaos Papageorgiou C, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. 2012;10(1):4–18. https://doi.org/10.2174/157016112798829760
- Filippi A, Constantin A, Alexandru N, et al. Integrins α4β1 and αVβ3 are reduced in endothelial progenitor cells from diabetic dyslipidemic mice and may represent new targets for therapy in aortic valve disease. Cell Transplant. 2020;29:1–8. https://doi.org/10.1177/0963689720946277
- Choi B, Shin M-K, Kim E-Y, et al. Elevated neuropeptide Y in endothelial dysfunction promotes macrophage infiltration and smooth muscle foam cell formation. Front Immunol. 2019;10:e1701. https://doi.org/10.3389/fimmu.2019.01701
- Singh S, Changkija S, Mudgal R, Ravichandiran V. Bioactive components to inhibit foam cell formation in atherosclerosis. Mol Biol Rep. 2022;49:2487–2501. https://doi.org/10.1007/s11033-021-07039-9
- Yari Z. Review of isoflavones and their potential clinical impacts on cardiovascular and bone metabolism markers in peritoneal dialysis patients. Prev Nutr Food Sci. 2022;27(4):347–353. https://doi.org/10.3746%2Fpnf.2022.27.4.347
- Libby P, Buring JE, Badimon L, et al. Atherosclerosis (Primer). Nat Rev Dis Prim. 2019;5:e56. https://doi.org/10.1038/s41572-019-0106-z
- Safian RD. Renal artery stenosis. Prog Cardiovasc Dis. 2021;65:60–70.
- Baran J, Kleczyński P, Niewiara Ł, et al. Importance of increased arterial resistance in risk prediction in patients with cardiovascular risk factors and degenerative aortic stenosis. J Clin Med. 2021;10(10):e2109. https://doi.org/10.3390/jcm10102109
- Gomez JA. Renin Angiotensin Aldosterone System Functions in Renovascular Hypertension. London, United Kingdom. Intechopen; 2021:79–116.
- Ma N, Wang SY, Sun YJ, Ren JH, Guo FJ. Diagnostic value of contrast-enhanced ultrasound for accessory renal artery among patients suspected of renal artery stenosis. Zhonghua Yi Xue Za Zhi. 2019;99(11):838–840. https://doi.org/10.3760/cma.j.issn.0376-2491.2019.11.008
- Akar EM, Aydın F, Tüzüner A, et al. Renal Autotransplantation in a Patient with Bilateral Renal Artery Stenosis Secondary to Takayasu Arteritis. Int J Organ Transplant Med. 2020;11(1):37–41.
- Stein EJ, Fearon WF, Elmariah S, et al. Left ventricular hypertrophy and biomarkers of cardiac damage and stress in aortic stenosis. J Am Heart Assoc. 2022;11(7):e023466. https:// doi.org/10.1161/JAHA.121.023466
- Nakagawa K, Nakashima Y. Pathologic intimal thickening in human atherosclerosis is formed by extracellular accumulation of plasma-derived lipids and dispersion of intimal smooth muscle cells. Atherosclerosis. 2018;274:235–242. https://doi.org/10.1016/j.atherosclerosis.2018.03.039
- Bobryshev Y V. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37(3):208–222. https://doi.org/10.1016/j.micron.2005.10.007
- Bec J, Vela D, Phipps JE, et al. Label-free visualization and quantification of biochemical markers of atherosclerotic plaque progression using intravascular fluorescence lifetime. Cardiovasc Imaging. 2021;14(9):1832–1842. https://doi.org/10.1016/j.jcmg.2020.10.004
- Xiang P, Blanchard V, Francis GA. Smooth muscle cell—macrophage interactions leading to foam cell formation in atherosclerosis: Location, location, location. Front Physiol. 2022;13:e921597. https://doi.org/10.3389/fphys.2022.921597
- Farahi L, Sinha SK, Lusis AJ. Roles of macrophages in atherogenesis. Front Pharmacol. 2021;12:e785220. https://doi.org/10.3389/fphar.2021.785220
- Yeo KP, Lim HY, Thiam CH, et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci Adv. 2020;6(50):eabc2697. https://doi.org/10.1126/sciadv.abc2697
- Yalçin F, Kucukler N, Cingolani O, et al. Evolution of ventricular hypertrophy and myocardial mechanics in physiological and pathological hypertrophy. J Appl Physiol. 2019;126(2):354–362. https:// doi.org/10.1152/japplphysiol.00199.2016
- Budzyń M, Gryszczyńka B, Boruczkowski M, et al. The potential role of circulating endothelial cells and endothelial progenitor cells in the prediction of left ventricular hypertrophy in hypertensive patients. Front Physiol. 2019;10:e1005. https://doi.org/10.3389/fphys.2019.01005
- Igbekele AE, Jia G, Hill MA, Sowers JR, Jia G. Mineralocorticoid receptor activation in vascular insulin resistance and dysfunction. Int J Mol Sci. 2022;23(16):e8954. https://doi.org/10.3390/ijms23168954
- Braam B, Lai CF, Abinader J, Bello AK. Extracellular fluid volume expansion, arterial stiffness and uncontrolled hypertension in patients with chronic kidney disease. Nephrol Dial Transplant. 2020;35(8):1393–1398. https://doi.org/10.1093/ndt/gfz020
- Lavenniah A, Luu TDA, Li YP, et al. Engineered circular RNA sponges act as miRNA inhibitors to attenuate pressure overload-induced cardiac hypertrophy. Mol Ther. 2020;28(6):1506–1517. https://doi.org/10.1016/j.ymthe.2020.04.006
- Bornstein AB, Rao SS, Marwaha K. Left Ventricular Hypertrophy. StatPearls Publishing; 2024.
- Poredos P, Poredos AV, Gregoric I. Endothelial dysfunction and its clinical implications. Angiology. 2021;72(7):604–615. https://doi.org/10.1177/0003319720987752
- Oliveira e Silva VR, Stringuetta Belik F, Hueb JC, et al. Aerobic exercise training and nontraditional cardiovascular risk factors in hemodialysis patients: results from a prospective randomized trial. Cardiorenal Med. 2019;9(6):391–399. https://doi.org/10.1159/000501589
- Chen Y, Freedman ND, Albert PS, et al. Association of cardiovascular disease with premature mortality in the United States. JAMA Cardiol. 2019;4(12):1230–1238. https://doi.org/10.1001/jamacardio.2019.3891
- Zhang D, Li L, Chen Y, et al. Syndecan-1, an indicator of endothelial glycocalyx degradation, predicts outcome of patients admitted to an ICU with COVID-19. Mol Med. 2021;27(1):e151. https://doi.org/10.1186/s10020-021-00412-1
- Barone Gibbs B, Hivert M-F, Jerome GJ, et al. Physical activity as a critical component of first-line treatment for elevated blood pressure or cholesterol: who, what, and how?: a scientific statement from the American Heart Association. Hypertension. 2021;78(2):e26–e37. https://doi.org/10.1161/HYP.0000000000000196
- Selva-O’Callaghan A, Alvarado-Cardenas M, Pinal-Fernández I, et al. Statin-induced myalgia and myositis: an update on pathogenesis and clinical recommendations. Expert Rev Clin Immunol. 2018;14(3):215–224. https://doi.org/10.1080/1744666X.2018.1440206
- Liu Z, Neuber S, Klose K, et al. Relationship between epicardial adipose tissue attenuation and coronary artery disease in type 2 diabetes mellitus patients. J Cardiovasc Med. 2023;24(4):244–252. https://doi.org/10.2459/JCM.0000000000001454
- Watts GF, Sullivan DR, Hare DL, et al. Integrated guidance for enhancing the care of familial hypercholesterolaemia in Australia. Hear Lung Circ. 2021;30(3):324–349. https://doi.org/10.1016/j.hlc.2020.09.943
- Moors J, Krishnan M, Sumpter N, et al. A polynesian-specific missense CETP variant alters the lipid profile. Hum Genet Genomics Adv. 2023;4(3):e100204. https://doi.org/10.1016/j.xhgg.2023.100204
- Rhainds D, Brodeur MR, Tardif J-C. Lipoprotein (a): when to measure and how to treat? Curr Atheroscler Rep. 2021;23:1–14. https://doi.org/10.1007/s11883-021-00951-2
- Tang Z, Li T, Peng J, et al. PCSK9: a novel inflammation modulator in atherosclerosis? J Cell Physiol. 2019;234(3):2345–2355. https://doi.org/10.1002/jcp.27254
- Yehuda H, E. LN. PCSK9 Inhibitors in lipid management of patients with diabetes mellitus and high cardiovascular risk: a review. J Am Heart Assoc. 2018;7(13):e008953. https://doi.org/10.1161/JAHA.118.008953
- Hong D-Y, Lee D-H, Lee J-Y, et al. Relationship between brain metabolic disorders and cognitive impairment: LDL receptor defect. Int J Mol Sci. 2022;23(15):e8384. https://doi.org/10.3390/ijms23158384
- A Romero C, Mathew S, Wasinski B, et al. Angiotensin‐converting enzyme inhibitors increase anti‐fibrotic biomarkers in African Americans with left ventricular hypertrophy. J Clin Hypertens. 2021;23(5):1008–1016. https://doi.org/10.1111/jch.14206
- Park S-J. The role of systolic blood pressure reduction in diastolic dysfunction: RAAS inhibition versus Non-RAAS blood pressure lowering. J Cardiovasc Imaging. 2020;28(3):183–185. https://doi.org/10.4250%2Fjcvi.2020.0083
- Pugliese NR, Masi S, Taddei S. The renin-angiotensin-aldosterone system: a crossroad from arterial hypertension to heart failure. Heart Fail Rev. 2020;25(1):31–42. https://doi.org/10.1007/s10741-019-09855-5