Environmental and Genetic Etiology of Rheumatoid Arthritis
Fazal Shan1, Muhammad Ibrahim Rashid2,Shah Faisal Jamal3, Muhammad Rahman4, and Irshad Ahmad1*
1Department of Molecular Biology and Genetics Institute of Basic Medical Sciences, Khyber Medical University, Peshawar, Pakistan
2Department of Biotechnology Virtual University of Pakistan
3Department Medical Lab Technology, Khyber Medical University, Peshawar, Pakistan
ABSTRACT
Background. Rheumatoid arthritis (RA) is a chronic inflammatory disorder that primarily affects joints, leading to pain, stiffness, and restricted mobility. Despite being idiopathic, multiple factors, including environmental influences, diet, microbiome alterations, and genetic predispositions, contribute to RA pathogenesis.
Methodology. A comprehensive review of recent studies was conducted to evaluate emerging molecular and cellular mechanisms involved in RA. The role of genetic and epigenetic factors in disease susceptibility and progression was analyzed.
Results. Several genetic factors, including HLA alleles and specific SNPs in genes such as PADI4, REL, RUNX1, FCGR2A, and CD40, have been implicated in RA susceptibility. Additionally, dysregulation of genetic molecules, including lncRNAs and miRNAs, along with inflammatory proteins such as IL-1 and CD28, contributes to disease progression. The interplay between immune responses, genetic predisposition, and environmental triggers plays a crucial role in RA pathogenesis.
Conclusion. Understanding the molecular and cellular mechanisms underlying RA can help identify novel therapeutic targets and enable the development of personalized treatment strategies for effective disease managementKeywords: environmental factors, epigenetic modification, genetic variations, HLA genes, infections, non-HLA gene, rheumatoid arthritis
- The crucial role of genetic elements, including HLA alleles and specific SNPs in key genes, in influencing susceptibility to rheumatoid arthritis (RA).
- The dysregulation of various genetic molecules, such as lncRNAs and miRNAs, and proteins, such as IL-1 and CD28, and their contribution to inflammation and disease progression in RA
- The current study highlights the potential implications of understanding these genetic mechanisms, suggesting that they could serve as targets for therapeutic interventions and contribute to the development of personalized treatment strategies for individuals with RA.
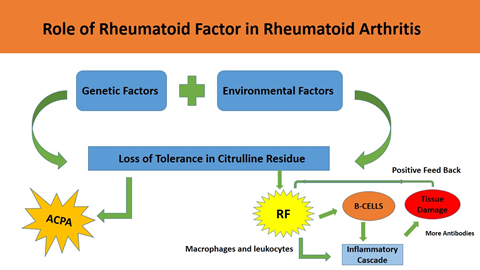
1. INTRODUCTION
Rheumatoid arthritis (RA) is an inflammatory disease that causes pain, stiffness, inflammation, and limited mobility in the joints. Over time, it can result in joint deformity, reduced functional ability, and decreased productivity, significantly affecting overall well-being of the patient. In advanced stages, the affected areas experience a decline in their normal functionality [1]. RA is an autoimmune condition that involves a complex interaction of cells and substances in the immune system. This interaction triggers the development of inflammation, both locally and throughout the body, at different disease stages [2].
The specific cause of RA is not fully known, although many aspects including infections, genetic variations, and mutations, have been linked to its development (Figure 1). Recent studies indicated that genomic mutations may contribute as a possible risk feature for the disease [3]. Furthermore, diverse environmental influences and lifestyle decisions, including dietary habits and levels of physical activity, have been noted to influence its onset and clinical features. Immune system components regulate tissue inflammation and restructuring, at both localized and systemic levels [4].
1.1. Contribution of Environmental Factors to RA
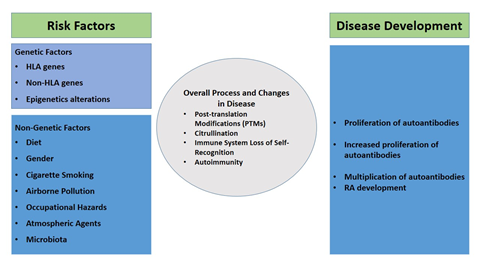
Environmental elements, such as nutrition, excess weight, tobacco use, air quality, and viral infections, trigger the growth of RA in individuals with genetic susceptibility (Table 1). Consequently, various studies have been conducted using both animal and human models to explore the causative molecular and cellular mechanisms, as well as possible targets for therapeutic interventions.
Higher environmental risk factors associated with RA are as follows: diet, obesity, airborne pollution, occupational hazards, atmospheric agents, cigarette smoking, and gut microbiota.
| Risk Genes for RA | Possible Causes and Mechanisms of RA |
|---|---|
| Major Histocompatibility Complex (MHC) genes HLA-DRB1 (01)(04) |
Enhances inflammation via citrullination due to citrulline antigens; prompts internal cellular changes |
| Peptidylarginine Deiminase Type 4 (PADI4) |
Enhances citrullination by converting arginine to citrulline |
| Ankyrin Repeat Domain 55 (ANKRD55) |
Gene intronic SNP (rs6859219) poses a risk for RA |
| Proto-oncogene, NF-kB Subunit (REL) |
SNPs in REL gene (e.g., rs13031237, rs13017599) associated with RA and increased citrullination |
| Runx Family Transcription Factor 1 (RUNX1) |
Modulates immune cell differentiation, activation, and functional regulation |
| FC gamma receptor (FCGR2A) | Variants (rs17400517, rs1801274, rs6671847, rs6668534) influence RA development |
| Nuclear Factor Kappa-B Kinase Subunit Epsilon (NFKBIE) | Variant rs2233434 (Val194Ala) affects cytokine expression and contributes to RA |
| Cytokines |
|
| Tumor Necrosis Factor Receptor (TNFR) Superfamily CD2, CD28, CD40 |
CD40 SNPs (rs1569723, rs1883832, rs4810485), CD209 variant (-96C>A) enhance cell activation and inflammation |
| DNA Methylation | Altered methylation patterns boost inflammation by changing protein production |
| RNAs | CircRNAs, sncRNAs (especially microRNAs), and lncRNAs regulate gene expression, inflammation, and immune responses; dysregulation contributes to RA progression |
1.1.1. DietThe role of diet in the occurrence and control of RA is considered significant. Specific dietary components, such as fiber, antioxidants, omega-3 fatty acids, and vitamins, exhibit anti-inflammatory characteristics and potential benefits in RA management. Conversely, certain dietary factors, such as saturated fats and processed sugars, may promote inflammation and contribute to the onset or worsening of RA [5]. Anti-oxidants make a vital contribution in diminishing inflammation and inhibiting the proliferation of cytokines and collagenase stimulated by TNF-α. It has been observed that RA-positive patients have insufficient levels of vitamin D. In some cases of RA, vitamin D supplementation is associated with disease chronicity and activity. Numerous studies have examined the potential influence of food on RA, while dietary modifications have been suggested as a supplementary strategy alongside conventional RA therapies [6]. Further investigation is essential to understand the precise dietary elements and mechanisms involved in RA development.
1.1.2. ObesityObesity is a well-recognized factor influencing the growth and advancement of RA. A recent meta-analysis further validates the development of this disease among overweight and obese individuals, as compared to those with normal body weight. In individuals with RA, obesity is linked to heightened disease activity, joint deterioration, and functional impairment [7]. The connection between obesity and the disease involves intricate and multifaceted mechanisms that are influenced by various factors [8]. Adipocytes play a key role amid obesity and RA. They stimulate systemic inflammation in RA by releasing inflammatory cytokines such as IL6, TNF-α, and leptin [9]. Additionally, adipose tissue releases free fatty acids that have the capacity to activate toll-like receptors (TLRs) and stimulate inflammation, further contributing to the association between obesity and RA [10]. Moreover, obesity can give rise to metabolic disturbances, including insulin resistance and dyslipidemia, which have been linked to a heightened susceptibility to the disease [11]. Insulin resistance and dyslipidemia also contribute to RA development by instigating the release of pro-inflammatory cytokines and also influence oxidative stress [12]. Obesity can also exacerbate joint stress and mechanical strain, particularly in weight-bearing joints, leading to joint damage and pain [13]. In addition, it can limit physical function and reduce the effectiveness of RA treatments (disease modifying antirheumatic drugs (DMARDs)) [14]. Overall, the facts suggest that obesity is a significant risk factor for RA. Addressing obesity through lifestyle interventions, such as diet and exercise, may have beneficial effects on disease outcomes [15]. Further investigation is essential to understand the role of obesity in RA, as well as to establish efficacious approaches to prevent and manage RA associated with obesity.
1.1.3. RA Etiology Linked to Airborne Pollution, Occupational Hazards, and Atmospheric Agents Numerous studies have explored the link between particulate pollutants and RA onset. Inhaling air particles stimulates the airways, activating the adaptive immune system. Inorganic dusts including asbestos and silica, as well as organic dusts such as animal and textile dusts, elevate RA risk. Male workers exposed to inorganic dusts show heightened susceptibility to both seropositive and seronegative RA, while exposure to organic dusts is associated with a higher susceptibility to seropositive RA. These effects may be attributed to bacterial endotoxins within organic dusts, activating immune response and inflammation in the airways [16]. The involvement of air pollutant in RA development has been confirmed using a combined model of collagen-induced arthritis (CIA) and increased airway inflammation prompted by organic dust extract (ODE). The combined model led to arthritis and bone loss, supporting the direct role of organic dusts in RA progression [17].
1.1.4. Cigarette Smoking (CS) Causes RA Several studies have identified air pollutants as a possible trigger for autoimmune disorders, such as RA, with the lungs considered as the initiation site for autoimmunity. The pathophysiological factor may involve systemic inflammation, oxidative stress, and airway damage, leading to immune responses. Among environmental factors, cigarette smoking (CS) is recognized as the most pertinent cause for developing RA [18]. A study on women confirmed that smoking plays a critical role in the disease's pathogenesis. It was the first to show that childhood exposure to tobacco smoke, even as a passive smoker, could increase RA risk. These findings support the theory that early external events could initiate the autoimmune response in RA development [19]. Smoking is a significant risk factor for this disease and various other disorders including lung and mouth carcinoma, pulmonary disorders, and cardiovascular conditions, along with the severity of RA pathogenesis, correlate with the intensity of smoking [20]. CS has been demonstrated to induce the generation of ACPA and RF, which lead to joint damage and bone loss, even during the early stages of the disease. However, this effect remains specific to the formation of ACPA, is not inclined via ethnicity, and is the outcome of the interface between smoking and HLA-DRB1-shared epitope (SE) alleles [21]. Individuals who smoke exhibit a dual occurrence of SE alleles, whereas non-smokers do not. Smoking has been found to enhance the expression of peptidylarginine deiminase (PAD) in the lungs. Consequently, it induces the conversion of arginine to citrulline in pulmonary peptide antigens [22]. Various studies have revealed that smoking RA patients show an elevated expression of the aromatic hydrocarbon receptor (AHR) and associated downstream genes in peripheral blood mononuclear cells (PBMCs), as compared to non-smoking RA patients. This implies that smoking might contribute to RA development by engaging the AHR pathway [23]. In a recent study, CS has been recognized as the predominant risk for triple-seropositivity, including RF, ACPA, and anti-carbamylated protein antibodies in RA patients. Specifically, it is associated with RA in individuals positive for one or two autoantibodies [24].
1.1.5. Association between Asthma and RA Recent case-control studies have proposed a direct link between mucosal surfaces, especially lung mucosa, and RA development. A strong association exists between clinically diagnosed asthma and RA, independent of confounding factors such as smoking intensity and duration. Inflammation and airway damage in asthma patients may cause protein citrullination and post-translational modifications (PTM), increasing ACPA production risk [25]. The observations indicate that respiratory mucosa makes a notable contribution to the pathogenesis of RA.
1.1.6. Role of Diet and Gut Microbiota in RA Development The potential involvement of microbiome in RA pathogenesis has been widely explored, as indicated by recent studies. During a metagenome-wide association study conducted in Japan, RA patients showed more Prevotella bacteria in their stool samples as compared to healthy individuals. RA samples had higher levels of the R6FCZ7 bacterial gene involved in redox reactions. These findings emphasize the crucial role of microbiome in RA development. Moreover, the findings also suggest potential therapeutic options in certain clinical trials [26]. A high-fat diet promotes pro-inflammatory gut microbiota. RA models and patients have reduced levels of short chain fatty acids (SCFAs) derived from gut microbes. Butyrate supplementation in CIA model improves disease activity by activating IL-10-producing Bregs, inhibiting plasmablast differentiation and autoantibody formation [27]. Furthermore, butyrate and larazotide acetate show benefits in disease treatment. Elevated levels of zonulin, a microbial protein that disrupts tight junctions, correlate with RA progression. An experiment showed that zonulin agonist worsens joint inflammation in CIA mice [28].
1.2. Role of Molecular Genetics in RA
Genetic abnormalities can lead to inherited disorders. Their investigation may provide valuable insights for researchers and clinicians to develop therapeutic interventions. In case of RA, it has been reported that 50–60% of the disease's susceptibility can be attributed to genetic heritability [29]. It is a common autoimmune arthritis, exhibiting a risk range of 0.3–1.1% in individuals of European descent and 0.1–0.5% in those of Asian heritage [30]. It has a strong heritability, with the HLA region of the MHC and non-MHC genes linked to the disease. GWAS identified genes including ILRK, IL35, IL12, CD40, CD209, FCRL, ADMATSL2, LRPA1, FCGR2A, NFKBIE, and PADI4 to be associated with increased susceptibility to RA (Table 2). These genes are crucial in regulating cytokine pathways and T cell proliferation, leading to auto-reactivity and autoimmune responses. Further analysis is required to enhance the comprehension of the disease's underlying mechanisms and to identify novel therapeutic avenues [31].
1.2.1. Role of Major Histocompatibility Complex (MHC) Genes in RA. Genetic variations in MHC genes, specifically HLA-DRB1 (04) and HLA-DRB1 (01) alleles make them susceptible to RA development. The existence of HLA-DRB1 (04:01) allele in RA patients is associated with anti-a501-515cit antibodies [32]. Concurrently, extensive investigation has been conducted regarding the key role of the HLA-B (08) allele in RA exposure, which has been linked specifically to serum positivity of anti-carbamylated protein antibodies [33]. HLA alleles have the potential to serve as predictive markers for therapeutic responses in RA patients, particularly in the context of biological agent treatments. The presence of the HLA DRB1(0404) allele might predict the response to anti-tumor necrosis factor (TNF) via alpha therapies in clinical practice.
1.2.2. Role of non-HLA Gene in RA
1.2.2.1. Peptidylargnine Deiminase Type 4 (PADI4)The PADI4 gene encodes for Peptidylarginine Deiminase Type 4 protein [34]. Citrullination, a process which converts arginine to citrulline within proteins, is not genetically encoded. Arginine deamination, facilitated by arginine deiminases (ADIs) and peptidylarginine deiminases (PADIs), leads to the production of citrulline, affecting protein structure and function and becoming a target for the immune system, thus contributing to autoimmune diseases including RA [35]. Citrullination and PADIs play an important role in RA pathophysiology. RA is characterized by the presence of antibodies to citrullinated proteins (ACPA), generated by PAD enzymes [36]. The PADI enzyme family, consisting of PADI1, 2, 3, 4, and 6, catalyze the citrullination process. Among these, PADI4 is particularly crucial in the pathogenesis of RA [37]. It encodes multiple isoforms that facilitate the post-translational citrullination of arginine in protein [38]. Recent research discovered single nucleotide polymorphisms (SNPs) in the exonic regions of the PADI4 gene, such as PADI4_89 (rs11203366), PADI4_90 (rs11203367), and PADI4_92 (rs874881), which have been linked to RA susceptibility. Several studies have also identified population-specific haplotypes associated with these SNPs [39].
1.2.2.2. Ankyrin Repeat Domain 55 (ANKRD55) Meta-analysis of GWAS indicates that the ANKRD55 gene intronic SNP (rs6859219) poses a risk for RA. Eukaryotic proteins containing ankyrin repeats—functional in transcription factors (TFs), cell cycle control, and other roles—can influence ANKRD55 gene regulation and protein expression, thereby affecting immune function and contributing to autoimmunity development [40].
1.2.2.3. Proto-oncogene, NF-kB Subunit (REL) The REL gene encodes the Proto-oncogene C-REL, a member of the RHD/IPT family, containing R.E.L homology and immunoglobulin-like fold and plexin domains [41]. This gene regulates inflammation, apoptosis, oncogenic processes, and immune responses. Mutations in REL are linked to RA development. It also plays a key role in B-cell proliferation and survival [42]. GWAS meta-analyses have identified SNPs such as rs13031237 and rs13017599 associated with RA susceptibility [43]. The rs13031237 SNP is further associated with anti-CCP positivity and RA risk.
1.2.2.4. RUNX1 (Runx Family Transcription Factor 1) RUNX1 is vital for cell production, embryonic development, lineage specification, differentiation, and apoptosis. It modulates immune cell differentiation and activation, influencing RA pathogenesis [44]. SNP rs9979383 has been significantly associated with RA in studies on the Chinese Han and Korean populations [45, 46].
1.2.2.5. Fc Gamma Receptor (FCGR2A) The FCGR2A gene encodes a receptor involved in recognizing immune complexes (ICs). It has emerged as a key contributor to RA pathogenesis [47]. GWAS have highlighted SNPs like rs17400517, rs1801274, rs6671847, and rs6668534 that are linked to RA susceptibility [48]. Analyzing these SNPs may help identify new therapeutic targets for RA.
1.2.2.6. NFKBIE (Nuclear Factor Kappa-B Kinase Subunit Epsilon) The NFKB pathway regulates cytokine expression in RA synovial tissue. GWAS meta-analyses identified non-synonymous SNPs in NFKBIE, including rs2233434 and Va1194A1a, associated with RA [49]. NFKBIE encodes a protein that binds NF-kappa-B complexes, sequestering them in the cytoplasm. Upon phosphorylation and subsequent ubiquitin-mediated degradation, NF-kappa-B is released to initiate gene expression in the nucleus [50]. This regulation plays a key role in RA inflammation and cytokine production.
1.2.2.7. ARID5B (AT-Rich Interaction Domain 5B) ARID5B, part of the ARID family (also known as MRF2/DESRT), acts as an epigenetic regulator by binding AT-rich DNA sequences. It forms a histone demethylase complex with PHD finger protein 2, impacting gene transcription during adipogenesis and liver development. SNP rs6479779 has been linked to both RA and autoimmune thyroid disease (AITD), suggesting shared genetic etiology [128, 129].
1.2.3. Role of Cytokines in RA Genetic variations in pro-inflammatory cytokines and cytokine receptor genes significantly contribute to RA susceptibility and development. IL6, a crucial cytokine in RA pathogenesis, is associated with specific SNPs (rs184229712, rs2069837, rs1800796, and rs36215814) in the IL6 gene, linking them to genetic susceptibility in recent investigations [51]. IL-1 cytokines, particularly IL-1α and IL-1β, play a significant role in RA pathogenesis by binding to IL-1 receptor 1 (IL-1R1). Recent research has linked three SNPs in the IL1R1 gene (rs3917318, rs956730, and rs1049057) with genetic predisposition to RA [52]. The gene encoding IL-1 R-associated kinase 1 (IRAK1) contributes to immune response stimulation. Several studies have indicated that the rs1059703 T allele of this gene is associated with RA development and correlates with disease severity. Dysregulated expression of IL-35, a new member of the IL-12 family, has been observed in RA. Recent investigations have identified an association between IL35 gene SNPs (rs2227314, rs2243131, rs9807813, and rs583911), indicating an elevated susceptibility to RA development in the Chinese population [53][54].
1.2.4. Tumor Necrosis Factor Receptor (TNFR) Superfamily Role in RA There has been an increasing focus on exploring novel genes encoding signaling regulatory molecules linked to RA development. The CD2 gene encodes a surface-associated protein on immune cells, interacting with CD58 on antigen-presenting cells, thereby promoting immune responses and acting as a co-stimulatory molecule on T and NK cells [55]. CD2, with roles in cell-cell adhesion, T cell activation, migration, cytokine production, and synovial tissue infiltration, emerges as a potential therapeutic target for RA. In RA, CD28, a key co-stimulatory receptor on T cells, plays a pivotal role in enhancing T cell activation, cytokine production, and also contributes to the autoimmune response and chronic inflammation typical of the disease. The association of CD28 genetic variants underscores its significance in RA pathogenesis [56]. CD40, a TNF family transmembrane glycoprotein expressed on various cells, serves as a potent T cell co-stimulatory factor. Recent studies have identified the association between specific CD40 gene polymorphisms (rs1569723, rs1883832, and rs4810485) and increased RA susceptibility, supported by co-dominant and dominant models. Additionally, the CD209 gene influences the immune response and controls monocyte-induced T cell activation in RA synovial tissue. The recently discovered -96C>A variant in the CD209 gene promoter is recognized as a potential genetic factor linked to RA [57][58].
1.2.5. Epigenetic Modifications in RA In RA pathogenesis, multiple molecular mechanisms are involved in the initiation and advancement of the disease. These include DNA methylation as well as the involvement of non-coding RNAs (ncRNAs) in significant capacities [59]. A comprehensive analysis of genome-wide association studies yielded an unexpected finding, indicating that roughly 80% of risk variants associated with RA are situated within non-coding regions of DNA [60]. Changes in gene expression, modifications of histones, alterations in chromatin structure, and the activity of transcription factors have a progressively important influence on RA development [61]. The recognition of the influence of epigenetic modifications on RA pathogenesis is increasing, as these modifications have the ability to regulate gene transcription, resulting in heritable alterations in phenotypes without modifying the underlying DNA sequence [62].
1.2.5.1. DNA Methylation in RA DNA methylation influences gene expression by altering DNA accessibility to transcription factors and regulatory proteins, predominantly targeting CpG dinucleotides, including those in CpG islands near gene promoters. Promoter hypermethylation induces a condensed heterochromatin state, inhibiting transcription factor binding and causing gene silencing. In contrast, promoter hypomethylation, with reduced methylation levels, fosters an open chromatin structure, facilitating active gene transcription [63]. DNA methylation significantly influences disease pathogenesis. In RA, distinctive aberrant DNA methylation patterns in specific genes, including IL6, IL10, and CXCL12, have been observed, leading to dysregulated gene expression and potentially contributing to disease susceptibility and activity [64]. Podgórska et al. explored the connection between DNA methylation of ADAMTSL2 and LRPAP1 genes and their impact on RA susceptibility and severity. ADAMTSL2 is linked to extracellular matrix remodeling, while LRPAP1 regulates specific signaling pathways. The study revealed that alterations in the methylation status of these genes can affect RA susceptibility and disease activity by modulating gene expression, influencing crucial biological processes and pathways in RA pathogenesis [59].
1.2.5.2. RNAs in RA Pathogenesis Circular RNAs (CircRNAs), small non-coding RNAs (sncRNAs), and long non-coding RNAs (lncRNAs) have also been implicated in the development of RA.
1.2.5.2.1. Circular RNAs (CircRNAs) Circular RNAs (circRNAs) are unique non-coding RNA transcripts with a circular structure formed through covalent binding of RNA ends, lacking 5' end caps or 3' poly A tails. Zheng et al. studied circRNA expression patterns in RA-positive individuals, identifying circRNAs such as hsa_circRNA_104194, hsa_circRNA_104593, hsa_circRNA_103334, hsa_circRNA_101407, and hsa_circRNA_102594 [65]. circMAPK9, a circular RNA, enhances pro-inflammatory actions in synovial fibroblasts (SF) by regulating the miR-140-3p/protein phosphatase magnesium-dependent 1A (PPM1A) axis. Additionally, circ_0088194, correlated with RA disease activity, interacts with miR-766-3p, leading to heightened expression of matrix metalloproteinase 2 (MMP2) and facilitating increased invasion and migration of SF in RA [66]. These discoveries indicate that circRNAs might contribute to the development of RA by potentially influencing the underlying mechanisms of the disease.
1.2.5.2.2. Small Non-coding RNAs (sncRNAs) sncRNAs, a subset of miRNAs, crucially regulate gene expression by binding to the 3' untranslated regions (3'UTRs) of target mRNAs. In RA, specific miRNAs such as miR-22-3p, 26b-5p, 142-3p, 155 show altered expression, potentially contributing to RA development by influencing genes associated with immune responses, inflammation, and tissue damage [67]. miRNA-22 has been consistently associated with RA in multiple studies. Treatment-naive RA patient serum exhibits both upregulation (hsa-miR-187-5p, -4532, -4516) and downregulation (hsa-miR-125a-3p, -575, -191-3p, -6865-3p, -197-3p, -6886-3p, -1237-3p, -4436b-5p) of various miRNAs, identified in diverse investigations. These findings highlight the potential role of these miRNAs in RA development and progression [68].
1.2.5.2.3. Long Non-coding RNAs (lncRNAs) lncRNAs, over 200 nucleotides long, lack protein-coding capacity, serve vital roles in gene regulation through diverse activities including chromatin remodeling, transcriptional control, RNA splicing, and post-transcriptional modifications, thus influencing development, disease, and cellular processes [69]. Several lncRNAs, including GAS5, are linked to RA pathogenesis. GAS5 exhibits reduced expression in fibroblast-like synoviocytes (FLS) and synovial tissues in RA patients. Conversely, Tanshinone IIA, a natural compound, has been found to elevate GAS5 expression without affecting the mRNA transcript and is known to reduce viability in FLS derived from RA patients [70]. lncRNAS56464.1 and linc00152 have been implicated in RA by influencing the proliferation of SF and modulating inflammatory responses, respectively [71]. They act as competitive endogenous RNAs, binding to miRNAs to disrupt their regulatory impact on target transcripts. For instance, lncRNA SNHG14 targets the miR-17-5p/MINK1 axis, promoting pro-inflammatory cytokine production and activating the JNK signaling pathway [72]. In another study, reduced plasma CASC2 levels were detected, correlating with elevated IL-17 production. Notably, a negative correlation between CASC2 and IL-17 was observed in both RA patients and healthy individuals. CASC2 expression promotes FLS apoptosis in RA individuals, suppressing IL-17 expression. These findings suggest the involvement of CASC2 downregulation in RA pathogenesis [73].
2. CONCLUSION
Rheumatoid arthritis (RA) is a complex autoimmune disease driven by a delicate interplay between genetic and environmental factors. Extensive study has unraveled the role of various genetic markers such as HLA alleles and SNPs in key genes including PADI4, REL, RUNX1, FCGR2A, and CD40, shedding light on RA susceptibility. Molecular factors, such as lncRNAs and miRNAs, and cytokines, such as IL-1 and CD28, play pivotal roles in RA pathogenesis. Understanding these genetic and molecular intricacies is essential for the development of personalized treatments, potentially improving patient outcomes. Moreover, it's crucial to recognize the substantial influence of environmental factors, such as diet and microbiome, in the development of RA. These aspects offer new insights for its prevention and treatment. A holistic approach that considers both genetic and environmental factors would pave the way for more effective management of the disease. This comprehensive review consolidates knowledge from diverse sources, providing a framework for future research and offering promise for better management and improved quality of life for individuals affected by RA
CONFLICT OF INTEREST
The authors of the manuscript have no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
DATA AVAILABILITY STATEMENT
Data availability is not applicable as no new data is collected.
FUNDING DETAILS
No funding has been received for this research.
REFERENCES
- Naqvi A, Hassali M, Aftab M, et al. Development of Evidence-Based Disease Education Literature for Pakistani Rheumatoid Arthritis Patients. Diseases. 2017;5(4):27. https://doi.org/10.3390/diseases5040027
- Giannini D, Antonucci M, Petrelli F, Bilia S, Alessia Alunno, Ilaria Puxeddu. One year in review 2020: pathogenesis of rheumatoid arthritis. Clinical and Experimental Rheumatology. 2020;38:387-397. https://doi.org/10.55563/clinexprheumatol/3uj1ng
- Radu AF, Bungau SG. Management of Rheumatoid Arthritis: an Overview. Cells. 2021;10(11):2857. https://doi.org/10.3390/cells10112857
- Carli L, Calabresi E, Governato G, Braun J, Reumatologia. One year in review 2018: axial spondyloarthritis. Clin Exp Rheumatol. 2019;37:889-898. https://i.clinref.com/data/uploads/articles/axial-spondyloarthritis-one-year-in-review-2018.pdf
- Häger J, Bang H, Hagen M, et al. The Role of Dietary Fiber in Rheumatoid Arthritis Patients: A Feasibility Study. Nutrients. 2019;11(10). https://doi.org/10.3390/nu11102392
- Dorji S, Yangchen S, chuki P. Prevalence of vitamin D deficiency in patients with autoimmune rheumatic diseases visiting the rheumatology clinic at the National Referral Hospital, Bhutan. SAGE Open Medicine. 2024;12. https://doi.org/10.1177/20503121231223313
- Slowikowski K, Wei K, Brenner MB, Raychaudhuri S. Functional genomics of stromal cells in chronic inflammatory diseases. Current Opinion in Rheumatology. 2018;30(1):65-71. https://doi.org/10.1097/bor.0000000000000455
- Lorenz H, Dalpke AH, Axel Deboben, et al. Mycobacterium kansasii tenosynovitis in a rheumatoid arthritis patient with long‐term therapeutic immunosuppression. Arthritis Care & Research. 2008;59(6):900-903. https://doi.org/10.1002/art.23717
- Pati S, Irfan W, Jameel A, Ahmed S, Shahid RK. Obesity and Cancer: a Current Overview of Epidemiology, Pathogenesis, Outcomes, and Management. Cancers. 2023;15(2):485. https://doi.org/10.3390/cancers15020485
- Metwaly A, Reitmeier S, Haller D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nature Reviews Gastroenterology & Hepatology. 2022;19. https://doi.org/10.1038/s41575-022-00581-2
- Hu Y, Costenbader KH, Gao X, et al. Sugar-sweetened soda consumption and risk of developing rheumatoid arthritis in women. The American Journal of Clinical Nutrition. 2014;100(3):959-967. https://doi.org/10.3945/ajcn.114.086918
- Tufvesson E, Bozovic G, Hesselstrand R, Bjermer L, Scheja A, Wuttge DM. Increased cysteinyl-leukotrienes and 8-isoprostane in exhaled breath condensate from systemic sclerosis patients. Rheumatology. 2010;49(12):2322-2326. https://doi.org/10.1093/rheumatology/keq271
- Tseng CC, Chen SN, Hwang JF, Lin CJ, Chen HS. Progressive outer retinal necrosis associated with occlusive vasculitis in acquired immunodeficiency syndrome. Journal of the Formosan Medical Association. 2012;114(5):469-472. https://doi.org/10.1016/j.jfma.2012.04.005
- Weng LH, Ko JY, Wang CJ, Sun YC, Wang FS. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis & Rheumatism. 2012;64(10):3267-3277. https://doi.org/10.1002/art.34602
- Cox AR, Masschelin PM, Saha PK, et al. The rheumatoid arthritis drug auranofin lowers leptin levels and exerts antidiabetic effects in obese mice. Cell Metabolism. 2022;34(12):1932-1946.e7. https://doi.org/10.1016/j.cmet.2022.09.019
- Ilar A, Gustavsson P, Wiebert P, Alfredsson L. Occupational exposure to organic dusts and risk of developing rheumatoid arthritis: findings from a Swedish population-based case–control study. RMD Open. 2019;5(2):e001049. https://doi.org/10.1136/rmdopen-2019-001049
- Poole JA, Thiele GM, K. Janike, et al. Combined Collagen‐Induced Arthritis and Organic Dust‐Induced Airway Inflammation to Model Inflammatory Lung Disease in Rheumatoid Arthritis. Journal of Bone and Mineral Research. 2019;34(9):1733-1743. https://doi.org/10.1002/jbmr.3745
- Gianfrancesco MA, Trupin L, Shiboski S, et al. Smoking Is Associated with Higher Disease Activity in Rheumatoid Arthritis: A Longitudinal Study Controlling for Time-varying Covariates. The Journal of Rheumatology. 2018;46(4):370-375. https://doi.org/10.3899/jrheum.180262
- Seror R, Henry J, Gusto G, Aubin HJ, Boutron-Ruault MC, Mariette X. Passive smoking in childhood increases the risk of developing rheumatoid arthritis. Rheumatology. 2018;58(7):1154-1162. https://doi.org/10.1093/rheumatology/key219
- Karlson EW, Chang S-C, Cui J, et al. Gene–environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Annals of the Rheumatic Diseases. 2009;69(01):54-60. https://doi.org/10.1136/ard.2008.102962
- Ishikawa Y, Ikari K, Hashimoto M, et al. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Annals of the Rheumatic Diseases. 2019;78(11):1480-1487. https://doi.org/10.1136/annrheumdis-2019-215463
- Murphy MP, Hunt D, Herron M, et al. Neutrophil-Derived Peptidyl Arginine Deiminase Activity Contributes to Pulmonary Emphysema by Enhancing Elastin Degradation. The Journal of Immunology. 2024;213(1):75-85. https://doi.org/10.4049/jimmunol.2300658
- Cheng L, Qian L, Xu ZZ, Tan Y, Luo CY. Aromatic hydrocarbon receptor provides a link between smoking and rheumatoid arthritis in peripheral blood mononuclear cells. PubMed. 2018;37(3):445-449.
- Regueiro C, Rodriguez-Rodriguez L, Lopez-Mejias R, et al. A predominant involvement of the triple seropositive patients and others with rheumatoid factor in the association of smoking with rheumatoid arthritis. Scientific Reports. 2020;10(1). https://doi.org/10.1038/s41598-020-60305-x
- Kronzer VL, Crowson CS, Sparks JA, Vassallo R, Davis JM. Investigating Asthma, Allergic Disease, Passive Smoke Exposure, and Risk of Rheumatoid Arthritis. Arthritis & Rheumatology. 2019;71(8):1217-1224. https://doi.org/10.1002/art.40858
- Yao Y, Cai X, Fei W, et al. Regulating Gut Microbiome: Therapeutic Strategy for Rheumatoid Arthritis During Pregnancy and Lactation. Frontiers in Pharmacology. 2020;11:594042. https://doi.org/10.3389/fphar.2020.594042
- Rosser EC, Piper CJM, Matei DE, et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metabolism. 2020;31(4):837-851.e10. https://doi.org/10.1016/j.cmet.2020.03.003
- Tajik N, Frech M, Schulz O, et al. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nature Communications. 2020;11(1). https://doi.org/10.1038/s41467-020-15831-7
- Song Y, Li J, Wu Y. Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders. Signal Transduction and Targeted Therapy. 2024;9(1). https://doi.org/10.1038/s41392-024-01952-8
- Kochi Y, Suzuki A, Yamada R, Yamamoto K. Ethnogenetic heterogeneity of rheumatoid arthritis—implications for pathogenesis. Nature Reviews Rheumatology. 2010;6(5):290-295. https://doi.org/10.1038/nrrheum.2010.23
- Yamamoto K, Okada Y, Suzuki A, Kochi Y. Genetic studies of rheumatoid arthritis. Proceedings of the Japan Academy, Series B. 2015;91(8):410-422. https://doi.org/10.2183/pjab.91.410
- Vetchinkina EA, Mikhaylenko DS, Kuznetsova EB, et al. Genetic Factors of Predisposition and Clinical Characteristics of Rheumatoid Arthritis in Russian Patients. Journal of Personalized Medicine. 2021;11(6):469. https://doi.org/10.3390/jpm11060469
- Regueiro C, Casares-Marfil D, Lundberg K, et al. HLA–B*08 Identified as the Most Prominently Associated Major Histocompatibility Complex Locus for Anti–Carbamylated Protein Antibody–Positive/Anti–Cyclic Citrullinated Peptide–Negative Rheumatoid Arthritis. Arthritis & Rheumatology. 2021;73(6):963-969. https://doi.org/10.1002/art.41630
- Mansouri P, Behmard E, Najafipour S, Kouhpayeh SA, Farjadfar A. Peptidylarginine deiminase (PAD): A promising target for chronic diseases treatment. International Journal of Biological Macromolecules. 2024;73(6):134576. https://doi.org/10.1016/j.ijbiomac.2024.134576
- Coenen D, Verschueren P, Westhovens R, Bossuyt X. Technical and Diagnostic Performance of 6 Assays for the Measurement of Citrullinated Protein/Peptide Antibodies in the Diagnosis of Rheumatoid Arthritis. Clinical Chemistry. 2007;53(3):498-504. https://doi.org/10.1373/clinchem.2006.078063
- Goldbach-Mansky R, Lee J, McCoy A, et al. Rheumatoid arthritis associated autoantibodies in patients with synovitis of recent onset. Arthritis Research. 2000;2(3):236-243. https://doi.org/10.1186/ar93
- Chang X, Xia Y, Pan J, Meng Q, Zhao Y, Yan X. PADI2 is significantly associated with rheumatoid arthritis. PloS One. 2013;8(12):e81259. https://doi.org/10.1371/journal.pone.0081259
- Perricone C, Ceccarelli F, Valesini G. An overview on the genetic of rheumatoid arthritis: A never-ending story. Autoimmunity Reviews. 2011;10(10):599-608. https://doi.org/10.1016/j.autrev.2011.04.021
- Baños-Hernández CJ, Navarro-Zarza JE, Parra-Rojas I, et al. PADI4 polymorphisms and the functional haplotype are associated with increased rheumatoid arthritis susceptibility: A replication study in a Southern Mexican population. Human Immunology. 2017;78(9):553-558. https://doi.org/10.1016/j.humimm.2017.05.005
- Li J, Mahajan A, Tsai MD. Ankyrin Repeat: A Unique Motif Mediating Protein−Protein Interactions. Biochemistry. 2006;45(51):15168-15178. https://doi.org/10.1021/bi062188q
- Ruiz-Perera LM, Greiner JFW, Kaltschmidt C, Kaltschmidt B. A Matter of Choice: Inhibition of c-Rel Shifts Neuronal to Oligodendroglial Fate in Human Stem Cells. Cells. 2020;9(4):1037. https://doi.org/10.3390/cells9041037
- Gilmore TD, Kalaitzidis D, Liang MC, Starczynowski DT. The c-Rel transcription factor and B-cell proliferation: a deal with the devil. Oncogene. 2004;23(13):2275-2286. https://doi.org/10.1038/sj.onc.1207410
- Salah E, Ahmed AA. A new piece of an old puzzle: lack of association between C-Rel (rs13031237-rs842647) single nucleotide polymorphisms and non-segmental vitiligo. Biomedical Dermatology. 2018;2(1). https://doi.org/10.1186/s41702-018-0027-6
- Samarakkody AS, Shin NY, Cantor AB. Role of RUNX Family Transcription Factors in DNA Damage Response. PubMed. 2020;43(2):99-106. https://doi.org/10.14348/molcells.2019.0304
- Yasmeen F, Pirzada RH, Ahmad B, Choi B, Choi S. Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies. International Journal of Molecular Sciences. 2024;25(14):7666. https://doi.org/10.3390/ijms25147666
- Dingwall HL. Developmental Genetics and the Evolution of Tendon Growth - ProQuest. ProQuest.com. Published 2019. https://www.proquest.com/openview/7dfa45728f0da12b28db04dd7a7c9fba/1?pq-origsite=gscholar&cbl=18750&diss=y
- Pandey JP. Genomewide Association Studies and Assessment of Risk of Disease. New England Journal of Medicine. 2010;363(21):2076-2077. https://doi.org/10.1056/nejmc1010310
- Fierabracci A, Arena A, Toto F, et al. Autoimmune polyendocrine syndrome type 1 (APECED) in the Indian population: case report and review of a series of 45 patients. Journal of Endocrinological Investigation. 2021;44(4):661-677. https://doi.org/10.1007/s40618-020-01376-5
- Yang Y, Peng L, He C, et al. The role of genetic variants in FCGR2A on the risk of rheumatoid arthritis in the Han Chinese population. Research Square. Published online September 2, 2020. https://doi.org/10.21203/rs.3.rs-63617/v1
- Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nature Reviews Immunology. 2016;16(4):247-258. https://doi.org/10.1038/nri.2016.9
- Chen J, Zhang A, Yang Y, Si Y, Hao D. Assessment of interleukin 6 gene polymorphisms with rheumatoid arthritis. Gene. 2020;765:145070. https://doi.org/10.1016/j.gene.2020.145070
- Liu X, Peng L, Li D, et al. The Impacts of IL1R1 and IL1R2 Genetic Variants on Rheumatoid Arthritis Risk in the Chinese Han Population: A Case–Control Study. International Journal of General Medicine. 2021;14:2147-2159. https://doi.org/10.2147/ijgm.s291395
- Xie Q, Xu WD, Pan M, et al. Association of IL-35 expression and gene polymorphisms in rheumatoid arthritis. International Immunopharmacology. 2021;90:107231. https://doi.org/10.1016/j.intimp.2020.107231
- Hu Z, Zhang L, Lin Z, et al. Prevalence and risk factors for bone loss in rheumatoid arthritis patients from South China: modeled by three methods. BMC Musculoskeletal Disorders. 2021;22(1). https://doi.org/10.1186/s12891-021-04403-5
- Matsui T, Connolly JE, Michnevitz M, et al. CD2 Distinguishes Two Subsets of Human Plasmacytoid Dendritic Cells with Distinct Phenotype and Functions. Journal of Immunology. 2009;182(11):6815-6823. https://doi.org/10.4049/jimmunol.0802008
- Binder C, Cvetkovski F, Sellberg F, et al. CD2 Immunobiology. Frontiers in Immunology. 2020;11. https://doi.org/10.3389/fimmu.2020.01090
- Huang Q, Xu WD, Su LC, Liu XY, Huang AF. Association of CD40 Gene Polymorphisms With Systemic Lupus Erythematosus and Rheumatoid Arthritis in a Chinese Han Population. Frontiers in Immunology. 2021;12. https://doi.org/10.3389/fimmu.2021.642929
- Chan HC, Wang SC, Lin CH, Lin YZ, Li RN, Yen JH. A novel CD209 polymorphism is associated with rheumatoid arthritis patients in Taiwan. Frontiers in Immunology. 2021;35(5). https://doi.org/10.1002/jcla.23751
- Podgórska D, Cieśla M, Majdan M, Podgórski R, Kolarz B. The relationship of ADAMTSL2 and LRPAP1 gene methylation level with rheumatoid arthritis activity. Clinical and Experimental Rheumatology. Published online August 4, 2021. https://doi.org/10.55563/clinexprheumatol/ogk9sd
- Doody KM, Bottini N, Firestein GS. Epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes. Epigenomics. 2017;9(4):479-492. https://doi.org/10.2217/epi-2016-0151
- Kolarz B, Podgorska D, Podgorski R. Insights of rheumatoid arthritis biomarkers. Biomarkers. Published online July 14, 2020:1-34. https://doi.org/10.1080/1354750x.2020.1794043
- Nemtsova MV, Zaletaev DV, Bure IV, et al. Epigenetic Changes in the Pathogenesis of Rheumatoid Arthritis. Frontiers in Genetics. 2019;10(570). https://doi.org/10.3389/fgene.2019.00570
- Moore LD, Le T, Fan G. DNA Methylation and Its Basic Function. Neuropsychopharmacology. 2012;38(1):23-38. https://doi.org/10.1038/npp.2012.112
- Klein K, Gay S. Epigenetic modifications in rheumatoid arthritis, a review. Current Opinion in Pharmacology. 2013;13(3):420-425. https://doi.org/10.1016/j.coph.2013.01.007
- Zheng F, Yu X, Huang J, Dai Y. Circular RNA expression profiles of peripheral blood mononuclear cells in rheumatoid arthritis patients, based on microarray chip technology. Molecular Medicine Reports. 2017;16(6):8029-8036. https://doi.org/10.3892/mmr.2017.7638
- Luo Z, Chen S, Chen X. CircMAPK9 promotes the progression of fibroblast-like synoviocytes in rheumatoid arthritis via the miR-140-3p/PPM1A axis. Journal of Orthopaedic Surgery and Research. 2021;16(1). https://doi.org/10.1186/s13018-021-02550-y
- Renman E, Brink M, Ärlestig L, Rantapää-Dahlqvist S, Lejon K. Dysregulated microRNA expression in rheumatoid arthritis families—a comparison between rheumatoid arthritis patients, their first-degree relatives, and healthy controls. Clinical Rheumatology. 2020;40(6):2387-2394. https://doi.org/10.1007/s10067-020-05502-9
- Cieśla M, Kolarz B, Majdan M, Darmochwał-Kolarz D. Plasma micro-RNA-22 is associated with disease activity in well-established rheumatoid arthritis. Clinical and Experimental Rheumatology. 2021;40(5). https://doi.org/10.55563/clinexprheumatol/zdhkrp
- Zhao Y, Vartak SV, Conte A, et al. “Stripe” transcription factors provide accessibility to co-binding partners in mammalian genomes. Molecular Cell. 2022;82(18):3398-3411.e11. https://doi.org/10.1016/j.molcel.2022.06.029
- Li G, Liu Y, Meng F, et al. Tanshinone IIA promotes the apoptosis of fibroblast-like synoviocytes in rheumatoid arthritis by up-regulating lncRNA GAS5. Bioscience Reports. 2018;38(5). https://doi.org/10.1042/bsr20180626
- Jiang H, Fan C, Lu Y, Cui X, Liu J. Astragaloside regulates lncRNA LOC100912373 and the miR 17 5p/PDK1 axis to inhibit the proliferation of fibroblast like synoviocytes in rats with rheumatoid arthritis. International Journal of Molecular Medicine. 2021;48(1). https://doi.org/10.3892/ijmm.2021.4963
- Zhang J, Lei H, Li X. LncRNA SNHG14 contributes to proinflammatory cytokine production in rheumatoid arthritis via the regulation of the miR-17-5p/MINK1-JNK pathway. Environmental Toxicology. 2021;36(12):2484-2492. https://doi.org/10.1002/tox.23361
- Liu C, Guo X, Bai S, Zeng G, Wang H. lncRNA CASC2 downregulation participates in rheumatoid arthritis, and CASC2 overexpression promotes the apoptosis of fibroblast like synoviocytes by downregulating IL 17. Molecular Medicine Reports. 2020;21. https://doi.org/10.3892/mmr.2020.11018