In Silico Characterization of Hypothetical Protein AZJ53_10480 in Streptococcus pneumoniae
Nimra Hanif1*, Sehrish Arshad2, Aqsa2, Muhammad Asim1, Amna Sadaqat Nadeem2, Tanzeel ur Rehman1, Nimra Shafique1, Raees Ahmad Khan1, and Moeez Manzor1
1Department of Biotechnology, Faculty of Science & Technology, University of Central Punjab, Lahore, Pakistan
2Riphah Institute of Clinical & Professional Psychology, Riphah International University, Lahore Pakistan.
Abstract
Background. Streptococcus pneumoniae is a major human pathogen responsible for serious infections such as pneumonia. Despite extensive research, many proteins in S. pneumoniae, including hypothetical proteins, remain uncharacterized, limiting the understanding of the bacterium's pathogenic mechanisms.
Methods. This study utilizes in silico tools to characterize the hypothetical protein AZJ53_10480 from S. pneumoniae. Sequence alignment and phylogenetic analysis were conducted using BLASTp and ClustalW, while PSIPRED and I-TASSER predicted the protein’s secondary and tertiary structures. Molecular docking studies were performed with AutoDock Vina to assess potential interactions with the antiviral drug sofosbuvir
Results. The in silico analysis revealed that the hypothetical protein AZJ53_10480 shares structural and functional similarities with viral capsid proteins of the hepatitis C virus. The protein was found to have a mixed localization, suggesting potential multifunctionality within the bacterial cell. Molecular docking studies indicated a strong binding affinity between AZJ53_10480 and sofosbuvir, suggesting that this protein could be a potential target for therapeutic intervention.
Conclusion. This study highlights structural properties and functional roles of hypothetical protein AZJ53_10480 in S. pneumoniae of . The findings suggest that AZJ53_10480 may play a role in the pathogenicity of this bacterium and could serve as a novel target for therapeutic development. Further experimental studies are needed to validate these findings and explore the protein's potential as a drug target.
Highlights
- In silico analysis reveals structural similarities between the hypothetical protein AZJ53_10480 and viral capsid proteins.
- Docking studies suggest AZJ53_10480 as a potential target for antiviral drugs like sofosbuvir.
- The study provides new insights into the possible pathogenic role of hypothetical proteins in Streptococcus pneumoniae.
Introduction
Streptococcus pneumoniae, or pneumococcus, is a gram-positive, circular, alpha-hemolytic (under high-impact conditions) or beta-hemolytic (under anaerobic conditions), facultative, anaerobic microorganism isolated from Streptococcus. These bacteria are non motile, do not form spores and are forund in pairs.It is a major cause of death in children, worldwide. As a critical human pathogenic bacterium, S. pneumoniae has been perceived as a significant cause of pneumonia since the late 19th century and remains the subject of numerous humoral resistances [1]. Pneumococcus spreads through respiratory droplets, such as those produced when an infected person coughs or sneezes. The bacteria can enter the body through the nose or mouth and then spread to the lungs, where it can cause pneumonia. It can also spread to other parts of the body, such as the bloodstream and brain. Pneumococcal infections can be serious, especially in young children, elderly adults, and people with weakened immune systems. In some cases, pneumococcal infections can lead to death [2].
In this study, a hypothetical protein (AZJ53_10480) was characterized in S. pneumoniae using in silico tools. Hence, this study provides information about the structure and function of hypothetical proteins. A comprehensive sequence analysis of the hypothetical protein was conducted, including the determination of its primary structure and the identification of conserved motifs. The study also investigated whether the hypothetical protein has any known associations with pathogenicity or virulence in S. pneumoniae, which may involve comparing it with proteins known to contribute in the pathogenicity of the bacterium. These objectives collectively aimed to provide a comprehensive understanding of the hypothetical protein AZJ53_10480 using in silico methods to uncover its sequence, structure, and potential biological functions.
There are multiple bacterial proteins that have different functions. In the respective bacteria, the proteins are particularly responsible for many important pathogenic activities, although some such proteins have yet to be characterized. Thus, protein characterization is required. Therefore, it remains unknown whether this protein is pathogenic, and if pathogenic, what the parameters are. If this protein is involved in any pathogenic activity, then there should be some drugs available to deal with it. .
In this study, the hypothetical protein AZJ53_10480 (S. pneumoniae) was utilized for in silico mutational analysis. The sequence of this protein was retrieved from NCBI and its structure was modeled and validated by using different types of tools including BLAST, CLUSTAL W, PSIPRED, Expasy protparam, I-TASSER, and CELLO . The modeled protein was then docked using AutoDock Vina [3].
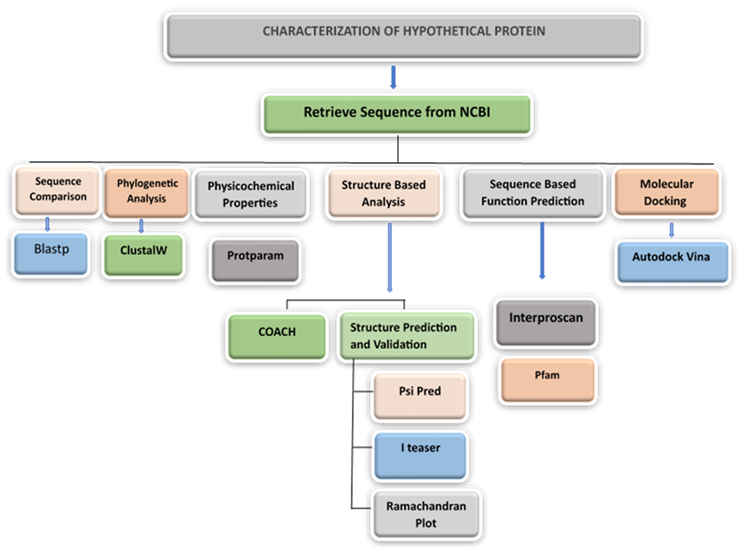
Figure 1. Layout of the methodology
MATERIALS AND METHODS
2.1. Data Collection
The genomic sequences of the hypothetical protein AZJ53_10480 were retrieved from the NCBI database. From the genome of S. pneumoniae, the hypothetical protein was selected having accession number TVX06118.1 and its sequence was retrived in FASTA format for futher analysis [4].
2.2. Sequence Analysis
The sequences were identified and aligned using BLASTp. It showed top hundred sequence results in four forms, namely description, graphic summary, alignments, and taxonomy. The top five proteins were selected for comparison. In graphics, the results appear to be based on score ranges that show the similarity between the sequences. The description shows the results on the basis of e-value, percentage identity, and query coverage. Further, alignments results give the number of matches between the sequences, the length of the sequences, and the mismatch or gaps collectively called identities. The authors downloaded the top ten sequences results. These results are shown in the description from BLAST. The Phymol bootstrap phylogenetic tree was generated using CLUSTALW [5].
2.3. Physiochemical Analysis
It was performed using ProtParam. Protparam performs a comprehensive analysis of protein sequences. It calculates various physiochemical properties of proteins and provides valuable insights into their structural and functional characteristics. These properties include amino acid composition, molecular weight, theoretical pI, extinction coefficient, instability index, aliphatic index, and the grand average of hydropathicity [6].
2.4. Secondary Structure Prediction
The secondary structure of the hypothetical protein AZJ53_10480 was predicted using PSIPRED. This is an Expasy tool that can predict protein function directly from amino acid sequences using limited or possibly no homology information. It uses artificial neural network and machine learning methods in its algorithms [7].
2.5. Tertiary Structural Prediction of Protein
Iterative reading assembly refinement (I-TASSER) was used to predict the tertiary structure and functions of protein. It provides sequence to sequence, sequence to structure, and structure to function analyses. In I TASSER, scores play an important role in predicting the efficiency of the model [8].
2.6. Domain Prediction of Protein
Domain and family similarity analyses were performed using InterPro software. This software predicts domains and classifies proteins into families. Conserved domains of AZJ53_10480 protein were predicted using CD NCBI [9].
2.7. Docking and Modeling with Ligand
Docking and modeling of the protein AZJ503_10480 were performed using AutoDock Vina and Patch Dock. The interaction between AZJ53_10480 and the ligand sofosbuvir was performed using AutoDock Vina and PatchDock. In PatchDock, protein and ligand were entered as input and the output comprised a list of potential complexes [10].
RESULTS
3.1. Phylogenetic Tree Construction
Protein BLAST was used to search for the alignment of this protein with other proteins that show similarities with it on the basis of description, alignment, and graphics. The top five protein sequences were obtained and a phylogenetic tree was constructed by using these FASTA sequences on ClustalW, as shown in Figure 2.
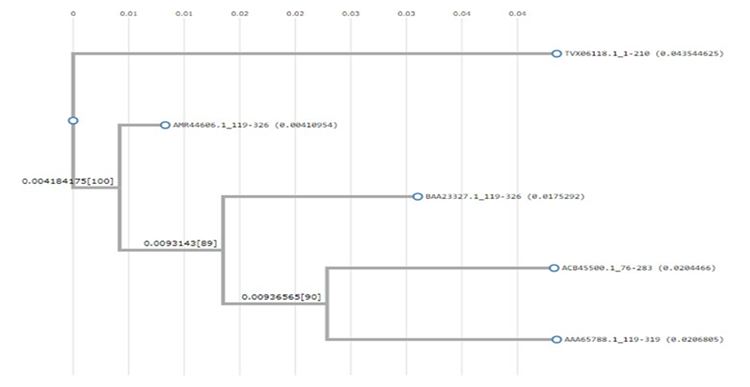
Figure 2. Phylogenetic Tree by ClustalW
This tree indicates similarity with some of the proteins but shows 100% similarity with WP_180958168.1, which is another hypothetical protein from a Streptococcus sp. of a different strain. It is also similar to the hepatitis C virus proteins in the coat. There are phylogenetic similarities between different proteins in the capsid and precursor proteins of hepatitis C viruses from different strains.
Table 1. Amino Acid Composition of Protein
|
Amino Acid Type |
No. of AA |
Percentage Composition |
|
Ala |
22 |
10.5% |
|
Arg |
9 |
4.3% |
|
Asn |
8 |
3.8% |
|
Asp |
9 |
4.3% |
|
Cys |
11 |
5.2% |
|
Gln |
5 |
2.4% |
|
Glu |
4 |
1.9% |
|
Gly |
22 |
10.5% |
|
His |
6 |
2.9% |
|
Ile |
10 |
4.8% |
|
Leu |
17 |
8.1% |
|
Lys |
2 |
1.0% |
|
Met |
7 |
3.3% |
|
Phe |
7 |
3.3% |
|
Pro |
11 |
5.2% |
|
Ser |
14 |
6.7% |
|
Thr |
12 |
5.7% |
|
Trp |
3 |
1.4% |
|
Tyr |
9 |
4.3% |
|
Val |
22 |
10.5% |
3.2. Physiochemical Analysis of Protein
ProtParam was used to perform a comprehensive analysis of physical and chemical properties of protein [9]. These pyscical and chemical properties include amino acid composition, molecular weight, theoretical pI, extinction coefficient, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). Table 1 shows the various types of amino acids and their percentage composition in the hypothetical protein AZJ53_10480. Grand average of hydropathicity (GRAVY) is 0.396. It was concluded that the proteins are hydrophilic in nature.
3.3. Secondary Structure Prediction by PSIPRED
PSIPRED showed the secondary structure of the hypothetical protein along with the colored regions and keys. It indicated that amino acid residues in the protein sequence contribute to the secondary structure, as shown in Figure 3. The figure shows the types of amino acids that make up the protein. Green residues are hydrophobic in nature, pink are polar amino acids, blue are aromatic amino acids, and small non-polar residues are shown in yellow [11, 12].

Figure 3. Amino Acid Types of Protein
3.4. Localization on Subcellular Level
MEMSAT is used for the subcellular localization of hypothetical proteins, their locations, and to determine whether the predicted components belong to the cellular region, extracellular region, or the membrane. The confidence score is provided as the reliability measure adjacent to the results. It confirms the accuracy of the predicted localization, as displayed in Figure 4.
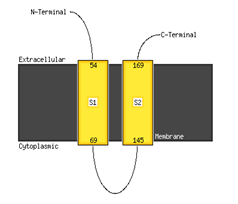
Figure 4. Localization of Protein Residues
Schematics for this hypothetical protein indicate that the yellow region in the figure indicates the extracellular region, the black region indicates the transmembrane helical region of the hypothetical protein, and the white region contributes to the cytoplasmic region of the protein.
3.5 Tertiary Structure Prediction via I-TASSER
I-TASSER only uses the templates of the highest significance in threading alignments, the significance of which is measured by Z-score, that is, the difference between the raw and average scores. The model with the best C-score, RMSD, and TM score was further confirmed and selected as the best predicted model on the basis of its parameters [13]. C-score of the model is -1.54, Z-score is 5.79, TM score is 0.33, and RMSD score is 13.9, as shown in Figure 5. On the basis of these scores, a protein model was selected and used.
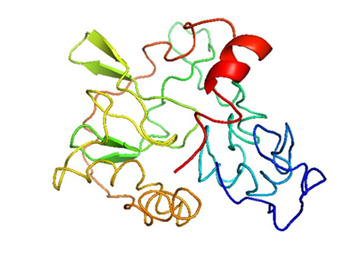
Figure 5. 3D Structure Predicted by I-TASSER
3.6. 3D Structure Validation of the Protein
A Ramachandran (RC) plot was used to validate the predicted protein structure. The RC plot displays the distribution of the φ and ψ angles for each residue in the protein structure. These angles define the rotation of peptide bonds which, in turn, determines the conformational geometry of the protein backbone. The x-axis represents the φ angle, which is the rotation around the Cα-N bond. The y-axis represents the ψ angle, which is the rotation around the Cα-C bond. The RC plot consists of three regions, that is, most allowed, allowed, and disallowed. The red area represents the most allowed, yellow represents allowed, and the white region represents disallowed [14]. In Figure 6, 91% of the amino acid residues are in the most allowed region which shows that the protein is good for further research.
|
Mode |
Affinity (kcal/mol) |
Dist from RMSD l. b |
Best Mode RMSD u.b. |
|
1 |
-5.7 |
0.000 |
0.000 |
|
2 |
-5.4 |
18.815 |
20.784 |
|
3 |
-5.4 |
2.445 |
3.590 |
|
4 |
-5.4 |
20.505 |
23.627 |
|
5 |
-5.4 |
12.377 |
15.731 |
|
6 |
-5.3 |
15.854 |
18.059 |
|
7 |
-5.3 |
8.433 |
12.195 |
|
8 |
-5.2 |
1.908 |
2.576 |
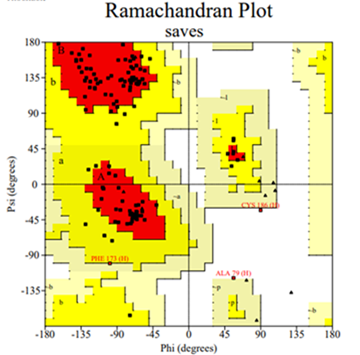
Figure 6. Protein Structure Validated by PROCHECK Program
3.7. Domain Prediction
InterPro was used to predict the domain and family of proteins. It showed close similarity to the viral capsid proteins in the hepatitis C virus. Pfam was used to predict the phylogenetic similarity of the protein domain [12]. It also showed significant similarity with the envelop proteins of the hepatitis C virus. For conserved domain analysis, CDNCBI server was used that also showed close similarity to this hypothetical protein. Therefore, it was confirmed that this protein is structurally and functionally related to the envelope protein of the hepatitis C virus.
3.8. Molecular Docking
The 3D structure of the protein was obtained through I-TASSER. The protein showed similarity with the proteins of hepatitis C virus. Therefore, a ligand sofosbuvir, which is effective against hepatitis C virus, was used to dock with the hypothetical protein AZJ53_10480, as mentioned in Table 2 and Figure 7.
Table 2. Affinity Energy from Docking with Ligand orientations
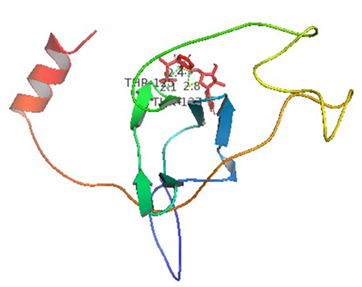
Figure 7. Protein Ligand Dock Complex Visualized by PyMOL
On docking, the first model was determined as the best model because it showed the best affinity, while the remaining models represented different orientations of the drugs to bind to the receptor protein in descending order of affinities. The determined affinity is better than the theoretical affinity. Therefore, the results showed a successful docking of the drug to the hypothetical protein. The docking results were visualized using PyMOL.
3.8.1. Atomic Interaction Analysis. The interaction between protein active sites and ligands was analyzed using PyMOL. Different amino acids showed hydrogen bonding with ligands at different positions. Threonine present at the position 125 showed ionic interactions with the ligand with a bond length of 2.4 angstrom and 2.1 angstrom. The results depicted the best interaction between the hypothetical protein AZJ53_10480 and sofosbuvir and can be used to design drugs against the said protein, as shown in Figure 8.
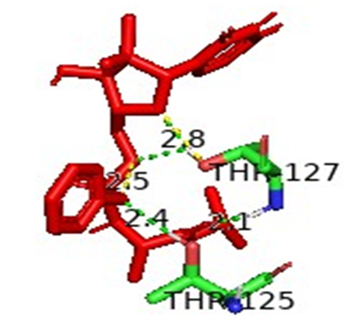
Figure 8. Analysis of Interaction between Active Sites and Ligand Binding Sites
DISCUSSION
This study was conducted to characterize a hypothetical protein AZJ53_10480 in S. pneumoniae using in silico tools. It aimed to determine the sequence, 2D and 3D structure, as well as the location of the protein in the cell, cellular or transmembrane regions, and at extracellular locations. The functions performed by the protein were also determined using I-TASSER. In order to find out ancestral relationships, homologies, and distant or closely related proteins, certain tools were used that indicated similarities with the proteins in the capsid of hepatitis C virus. Manual and automated docking via AutoDock Vina and Patch Dock were performed by using the ligand Sofosbuvir that is effective against viral proteins. The affinities were found to be quite useful, indicating higher binding affinity as compared to theoretical values. Although the characterization of protein has been achieved in this research, certain impurity has been identified as well within the protein. Hence, it should be refined in vitro in order to completely accept its functions and submission.
This study aligns with previous research on the characterization of hypothetical proteins in various bacterial species, such as Streptococcus pneumoniae [15], Streptococcus mutans [16] and Shigella dysenteriae [17]. These studies emphasized the importance of elucidating the functions of hypothetical proteins to understand bacterial pathogenesis and identify potential drug targets. Additionally, the use of in silico tools for protein characterization is supported by research on protein-DNA interactions [18], annotation of uncharacterized proteins [19], and advances in protein complex analysis [20], highlighting the significance of computational approaches in modern proteomics research.
Moreover, the purification and refinement of the hypothetical protein AZJ53_10480 can benefit from techniques such as biotinylation for efficient purification [21], affinity chromatography for one-step purification of recombinant proteins [22], and tangential flow filtration for novel purification methods [23]. These methods can aid in obtaining high purity proteins for further experimental validation and functional studies. Also, in silico characterization of the hypothetical protein AZJ53_10480 from S. pneumoniae provides valuable insights into its structure, potential functions, and interactions. Further experimental validation and purification are essential to confirm its role and potential applications, especially in the context of developing novel therapeutics against bacterial infections.
4.1. Conclusion
This study carried out the in silico characterization of a hypothetical protein AZJ53_10480 in S. pneumoniae using mutation analysis tools. To check the mutational effect on the biological function of the protein, PROVEAN was used. Using different tools for photochemical properties, subcellular localization, structure, and domain prediction, conserved region mutation analysis of the protein was performed. PSIPRED results showed that the amino acids present in the protein sequence contribute to the secondary structure. PSIPRED cartoons in the form of peaks also showed accuracy because a higher peak indicates a more confident structure. The results of manual docking by AutoDock Vina showed successful docking to the hypothetical protein.
Conflict of Interest
The author of the manuscript has no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
Data Availability Statement
The data associated with this study will be provided by the corresponding author upon request.
Funding details
This research did not receive grant from any funding source or agency.
Bibliography
- Appelbaum PC. Antimicrobial resistance in streptococcus pneumoniae: an overview. Clinic Infect Dis. 1992:15(1):77–83. https://doi.org/10.1093/clinids/15.1.77
- Lynch JP, Zhanel GG. Streptococcus pneumoniae: epidemiology, risk factors, and strategies for prevention. Semin Respir Crit Care Med. 2009;30(2):189–209. https://doi.org /10.1055/s-0029-1202938
- Rabbi MF, Akter SA, Hasan MJ, Amin A. In silico characterization of a hypothetical protein from Shigella dysenteriae ATCC 12039 reveals a pathogenesis-related protein of the type-VI secretion system. Bioinf Biol Insights. 2021;15:1–12. https://doi.org/10.1177/11779322211011140
- Ahmed F, Ahmed N, Prome AA, Robin TB, Rani NA. In silico identification of Shigella sonnei hypothetical protein RUK71877. 1 as interleukin receptor mimic Protein A and a potential drug target. Int J Biosci. 2022;21(6);7–17.
- Tasneem M, Gupta SD, Momin MB, Hossain KM, Osman TB, Rabbi MF. In silico annotation of a hypothetical protein from Listeria monocytogenes EGD-e unfolds a toxin protein of the type II secretion system. Genom Inform. 2023;21(1):e7. https://doi.org/10.5808%2Fgi.22071
- Shahrear S, Zinnia MA, Sany MR, Islam AB. Functional analysis of hypothetical proteins of vibrio parahaemolyticus reveals the presence of virulence factors and growth-related enzymes with therapeutic potential. Bioinf Biol Insights. 2022;16:1–16. https://doi.org/10.1177/11779322221136002
- Rabbi MF, Akter SA, Hasan MJ, Amin A. In silico characterization of a hypothetical protein from ATCC 12039 reveals a pathogenesis-related protein of the type-VI secretion system. Bioinf Biol Insights. 2021;15:1–12. https://doi.org/10.1177/ 11779322211011140
- Masum MH, Rajia S, Bristi UP, et al. In silico functional characterization of a hypothetical protein from pasteurella multocida reveals a novel s-adenosylmethionine-dependent methyltransferase activity. Bioinf Biol Insights. 2023;17:1–17. https://doi.org/10.1177/11779322231184024
- Munna MM, Islam MA, Shanta SS, Monty MA. Structural, functional, molecular docking analysis of a hypothetical protein from Talaromyces marneffei and its molecular dynamic simulation: an in-silico approach. J Biomol Struc Dyn. 2024:1–20. https://doi.org/10.1080/07391102.2024.2314264
- Morris GM, Lim-Wilby M. Molecular docking. In: Kukol A, ed. Molecular Modeling of Proteins. Springer; 2008:365–382. https://doi.org/10.1007/978-1-59745-177-2_19
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. https://doi.org/10.1093/bioinformatics/16.4.404
- Liu J, Rost B. Sequence-based prediction of protein domains. Nucl Acids Res. 2004;32(12):3522–3530. https://doi.org/10.1093/nar/gkh684
- Mondol SM, Das D, Priom DM, Rahman MS, Islam MR, Rahaman MM. In silico identification and characterization of a hypothetical protein from rhodobacter capsulatus revealing s-adenosylmethionine-dependent methyltransferase activity. Bioinf Biol Insights. 2022;16:1–16. https://doi.org/10.1177/11779322221094236
- Laskowski RA, Furnham N, Thornton JM. The ramachandran plot and protein structure validation. Biomol Form Func. 2013;62–75. https://doi.org/10.1142/9789814449144_0005
- Jiang YL, Zhang JW, Yu WL, et al. Structural and enzymatic characterization of the streptococcal ATP/Diadenosine polyphosphate and phosphodiester hydrolase Spr1479/SapH. J Biol Chem. 2011;286(41):35906–35914.
- Nan J, Brostromer E, Liu XY, Kristensen O, Su XD. Bioinformatics and structural characterization of a hypothetical protein from streptococcus mutans: implication of antibiotic resistance. PloS One. 2009;4(10):e7245. https://doi.org/10.1371/journal.pone.0007245
- Rabbi MF, Akter SA, Hasan MJ, Amin A. In silico characterization of a hypothetical protein from<i>Shigella Dysenteriae</I>ATCC 12039 reveals a pathogenesis-related protein of the Type-Vi secretion system. Bioinf Biol Insights. 2021;15:1–12. https://doi.org/10.1177/11779322211011140
- Cozzolino F, Iacobucci I, Monaco V, Monti M. Protein–DNA/Rna interactions: an overview of investigation methods in the -omics era. J Prot Res. 2021;20(6):3018–3030. https://doi.org/10.1021/acs. jproteome.1c00074
- Ijaq J, Chandrasekharan M, Poddar R, Bethi N, Sundararajan VS. Annotation and curation of uncharacterized proteins- challenges. Front Genet. 2015;6:e119. https://doi.org/10.3389/fgene.2015.00119
- Gingras AC, Aebersold R, Raught B. Advances in protein complex analysis using mass spectrometry. J Physiol. 2005;563(1):11–21. https://doi.org/10.1113/jphysiol.2004.080440
- Fairhead M, Howarth M. Site-Specific biotinylation of purified proteins using BirA. In: Gautier A, Hinner MJ, eds. Site-Specific Protein Labeling. Methods in Molecular Biology. Humana Press; 2014: 171–184. https://doi.org/10.1007/978-1-4939-2272-7_12
- Verma V, Kaur C, Grover P, Gupta A, Chaudhary VK. Biotin-tagged proteins: reagents for efficient ELISA-based serodiagnosis and phage display-based affinity selection. PloS One. 2018;13(1):e0191315. https:// doi.org/10.1371/journal.pone.0191315
- Wang HZ, Chu ZZ, Chen CC, et al. Recombinant passenger proteins can be conveniently purified by one-step affinity chromatography. PloS One. 2015;10(12):e0143598. https://doi.org/10.1371/journal.pone.0143598