
| Review | Open Access |
|---|
Exploring the Potential Anti-diabetic Properties of Rhapis excelsa through Computational Approaches |
 |
|---|
![]() Iqra Sagheer, Sania Riaz* , Mahnoor Azhar, Mariam Tariq, Hamza Masood, and Sharjeel Tariq
Iqra Sagheer, Sania Riaz* , Mahnoor Azhar, Mariam Tariq, Hamza Masood, and Sharjeel Tariq
Department of Bioinformatics and Biosciences, Faculty of Health and Life Sciences, Capital University of Science and Technology, Islamabad, Pakistan
Background. Phytotherapy has been practiced against many acute and chronic diseases by different ethnic groups around the globe. Among these medicinal plants, the species of Arecaceae family stands out due to their vast economic importance. Most of the family members contain rich phytochemicals and secondary metabolites, being part of this family, Rhapis excelsa contains flavonoids, terpenoids, tannins, and many other bioactive metabolites which are believed to contribute to its medicinal properties, potentially offering therapeutic benefits against Diabetes mellitus which is a complex metabolic disorder that has become a major global health concern. The aim was to identify and select the potential bioactive compounds from Rhapis excelsa by using computational approaches for the treatment and management of DM.
Methods. C-reactive protein (CRP) was selected as the target protein. Three different ligands from Rhapis excelsa underwent both physicochemical and toxicity assessments using SwissADME and ProTox-II. Computational approaches, such as molecular docking and virtual screening were used to evaluate binding affinity.
Results. The leading compound identified was apigenin which was then compared to the standard anti-diabetic drug, Glibenclamide. The results indicated that apigenin and Glibenclamide have similar functions when selected as ligands against the target protein. Literature evidence also supports the anti-diabetic effects of apigenin.
Conclusion. The current study identified potential phytochemical compounds from Rhapis excelsa using computational approaches for their application in diabetes drug development. Computational analyses including molecular docking and virtual screening revealed some bioactive compounds within Rhapis excelsaGRAPHICAL ABSTRACT

1. INTRODUCTION
Medicinal plants are crucial in managing Diabetes mellitus (DM), a serious metabolic condition. Traditional herbs are known to possess significant anti-diabetic properties without adverse side effects [1]. These plants play an essential role in healthcare, particularly in managing various infectious diseases due to their abundance of bioactive phytochemicals. Recently, there has been a growing global focus on medicinal plant-based therapies to discover affordable and safe treatments for diabetes [2]. Over the last ten years, there has been a notable rise in the use of complementary and alternative medicines (CAM). These include medicinal herbs and dietary supplements, for managing chronic illnesses, including diabetes [3]. Herbal medicines are favored for their effectiveness to treat various diseases and disorders. This is because they are generally associated with fewer side effects, are readily available, and are cost-effective [4].
Diabetes is a chronic non-communicable disease (CNCD) characterized by chronic hyperglycemia due to deficiencies in the release and function of the pancreatic hormone insulin. This condition falls under the category of metabolic disorders and represents a significant global health concern. Additionally, diabetes may result from pancreatic damage and some rare inherited forms. Early diagnosis and appropriate management of blood sugar level, blood pressure as well as cholesterol levels could prevent or delay the complications that are associated with diabetes. The prevalence of diabetes is notably higher in Asian countries, especially China and India [5]. According to global statistics, approximately 463 million adults are affected, with 90% of them suffering from type 2 DM [6]. According to an article by "The News", Pakistan ranks third worldwide in diabetes prevalence, following China and India [7]. In Pakistan, the prevalence of diabetes was 11.77% in 2016 [8], 16.98% in 2018 [9], and 17.1% in 2019 [7], [8].
Flavonoids, a large class of plant-derived secondary metabolites, are characterized by their polyphenolic structures. These can be classified into six primary groups: flavonols, flavan-3-ols (flavanols), flavones, anthocyanins, isoflavones, and flavanones [9]. Among these, flavones, such as luteolin and apigenin are particularly prevalent. Scientific research has exclusively explained the health benefits of flavonoids that highlights their extensive pharmacological properties including anti-inflammatory, anti-microbial, anti-diabetic as well as anti-tumorigenic effects [10]. Flavonoids also play a crucial role in the management of metabolic disorders, such as cardiovascular diseases (CVDs), cancer, obesity, and diabetes. Most importantly, their anti-diabetic properties facilitate in regulating carbohydrate digestion in the body, insulin signaling and secretion, glucose uptake and fat storage as well [10]. Apigenin has been recognized for its dual role as an antioxidant and an anti-diabetic agent in several computational and experimental studies [11].
The Angiosperm family, Arecaceae, commonly known as the palm family, comprises approximately 3,000 species across 200 genera. Among these species, there is the decorative plant Rhapis excelsa, commonly referred to as lady palm or bamboo palm. This species is marked by its palmate leaves and is commonly cultivated in pots. Due to its dense and bushy growth, Rhapis excelsa is also known as a hedge plant. In China, the canes of Rhapis excelsa are used as walking sticks and umbrella handles. The dried, fibrous leaf stalk bases of plants are used in traditional Chinese medicine. Furthermore, the ashes of its burned bark are supposed to enhance blood circulation and are used in the treatment of rheumatism. These ashes are also applied externally to help control bleeding. The chromatographic analysis of the leaves of this plant has revealed the existence of four flavonoids: vitexin, vicenin-2, isoorientin, and orientin [12].
C-reactive protein (CRP) is a well-established positive acute-phase reactant that is identified as a key factor in the inflammatory response of the body. It is primarily produced by hepatocytes and excreted into the bloodstream during different episodes of inflammation. Furthermore, this act as a reliable biomarker for numerous inflammatory conditions and diseases [13]. It is characterized as a serum protein that has the ability to precipitate the C-polysaccharide of the pneumococcal cell walls in the presence of calcium and has a role in the acute phase of infection. CRP is a highly conserved protein, belonging to the pentraxin family and is found across species ranging from arthropods to humans. The structure of CRP contains a 206 amino-acid cyclic pentamer that consists of five identical subunits connected non-covalently, with molecular weight of approximately 23 kDa. Each subunit of CRP forms a discoid shape having a central pore and features two antiparallel, double-layered β sheets. Its elevated levels have been strongly linked with chronic inflammation associated with Type 2 Diabetes Mellitus (T2DM). In human beings, it plays a critical role during the acute phase response in the body for inflammation, infection, and tissue injury, with levels increasing dramatically up to 1000-fold within 24 to 72 hours following the inception of these conditions. Chronic hyperglycemia, that is a hallmark of diabetes, aggravates this process by creating oxidative stress and inflammation, causing both local and systemic damage. Many studies have shown a significant correlation between elevated CRP levels with the development of T2DM, even after accounting for other risk factors, such as obesity, hyperinsulinemia, hypertriglyceridemia as well as low HDL cholesterol [14].
Therefore, targeting CRP could offer a novel therapeutic approach to managing T2DM-associated inflammation. The expression of CRP is regulated by interleukin 6 (IL-6) as well as TNF-α, secreted by adipocytes [10].
2. MATERIALS AND METHODS
2.1. Retrieval of 3D Structure of Target ProteinThe structural elucidation of the target protein is an essential preliminary step in molecular docking and virtual screening studies. For this study, C-reactive protein (CRP), a well-established biomarker of systemic inflammation and a key player in type 2 diabetes mellitus (T2DM) progression, was selected. The three-dimensional structure of CRP was required for docking analysis with selected phytoconstituents of Rhapis excelsa. Due to the unavailability of a complete experimental structure with sufficient resolution in the Protein Data Bank (PDB), a homology modeling approach was employed using the CRP amino acid sequence.
2.1.1. Primary Sequence of Target Proteins. The primary sequence of targeted proteins CRP was taken in FASTA format using UniProt database (http://www.uniprot.org/) under accession numbers P02741 with 224 residue lengths.
2.1.2. Retrieval of Protein Structure. The CRP sequence obtained from Uniprot was employed for protein structure homology modelling using Swiss-Model (https://swissmodel.expasy.org/).
2.1.3. Analysis of Physicochemical Properties of Protein. The physicochemical properties of any proteins are crucial in defining their functions. So, the physicochemical parameters of CRP were determined using the ProtParam tool (https://web.expasy.org/protparam/).
CRP was selected as the target protein. This is because elevated CRP levels are closely linked to the pathogenesis of (T2DM). Its central role in chronic inflammation and metabolic dysfunction makes it a suitable biomarker and therapeutic target in diabetes-related studies.
2.2. Retrieval of Ligands Structure and ADMET Evaluation2.2.1. 3D Structure of Ligands. The ligands utilized in this study were obtained from PubChem database and consist of the chemical compounds found in Rhapis excelsa (https://pubchem.ncbi.nlm.nih.gov/).
However, the 3D structures of the ligands were not displayed in Table 2 and must be added or referenced appropriately in the final version as per reviewer suggestions.
2.2.2. ADMET Analysis. Furthermore, the screening of the selected ligands was done by Swiss ADME (http://www.swissadme.ch/) to estimate physicochemical descriptors. These additionally provide predictions for ADME parameters, characteristics for pharmacokinetic feature, druglike traits, and medicinal chemistry compatibility for ligands, with the aim of facilitating drug discovery initiatives. Subsequently, an examination of toxicity was carried out using the Protox-2 (https://tox-new.charite.de/).
2.3. Ligand-receptor Molecular Docking2.3.1. Molecular Docking. PatchDock (http://bioinfo3d.cs.tau.ac.il/PatchDock/) is a software commonly employed to conduct molecular docking experiments. In this study, PatchDock was utilized to investigate the docking interactions between the CRP and the phytoconstituents found in Rhapis excelsa. The selection of docking results is based on the highest docking score. PatchDock is a geometry-based molecular docking algorithm designed to identify docking transformations with good molecular shape complementarity. Furthermore, the identification of the active site of a target protein is facilitated through the utilization of Castp (http://sts.bioe.uic.edu/), an online tool that is readily accessible. The study provided predictions regarding the locations suitable for binding as well as an analysis of the surface area and volume of protein pockets.
2.3.2. Standard Drug Docking with Target Protein. Glibenclamide was selected as the standard drug for docking. The results obtained were compared with the docking outcomes of ligands. Additionally, the ADME characteristics and toxicity of glibenclamide were assessed. Furthermore, the interaction of standard drugs with target protein was compared with Protein-ligand interaction.
3. RESULTS
3.1. Protein Structure Modeling and Physicochemical AnalysisPrimary sequence of CRP was retrieved in FASTA format “Figure 1” from the UniProt database (http://www.uniprot.org) using the accession number P02741.

Figure 1. Amino Acid Sequence of CRP
3.1.1. Homology-Modelling. The protein sequence of CRP, obtained from the Uniprot database, was utilized for the purpose of 3D-protein structure homology modelling through Protein Data Bank (https://www.rcsb.org).

Figure 2. 3D Model of CRP Structure Obtained through Homology Modeling
The physicochemical properties of CRP were determined through the use of the ProtParam (https://web.expasy.org/protparam/), an online tool. The web application was utilized to compute several physical and chemical properties of the protein. The computed parameters encompassed several parameters, such as theoretical isoelectric point (pI), composition of amino acid with regards to positive and negative charge, and atomic compound.
The physiochemical analysis in Table 1 reveals that the protein's molecular weight is 24925.42. The isoelectric point (pI) of CRP is determined to be 5.45. There exists a higher number of positively charged amino acids in comparison to negatively charged amino acids. Accordingly, extinction coefficient 1 corresponds to a value of 45045 M-1 cm-1, while the extinction coefficient 2 corresponds to a value of 44920. The instability index of CRP is calculated to be 38.04. Furthermore, the aliphatic index was determined to be 83.41, while the GRAVY score demonstrated a value of -0.052.
Table 1. Physiochemical Properties of CRP
|
Molecular weight |
pI |
Negatively Charged Residues |
Positively Charged Residues |
Ext. coefficient 1 |
Ext. coefficient 2 |
Instability Index |
Aliphatic Index |
Gravy |
|---|---|---|---|---|---|---|---|---|
|
24925.42 |
5.45 |
24 |
20 |
45045 |
44920 |
38.04 |
83.41 |
-0.052 |
3.1.2. Binding Affinity and Active side Determination. PatchDock (http://bioinfo3d.cs.tau.ac.il/PatchDock/) is a computational tool used to perform molecular docking between a target protein and apigenin. Upon executing the PatchDock algorithm, the file exhibiting the highest docking score was retrieved for further assessment. The highest docking score obtained was 4440.
The ligand exhibits the highest affinity for the protein when it binds to the active site of the target protein. In order to get results pertaining to the active site of CRP, Castp (http://sts.bioe.uic.edu/) was utilized.

Figure 3. Active Site of CRP Shown with Red Pocket Region. Binding Pocket Area = 24263.303 Ų; Volume = 53142.377 ų.
The red colored region (Figure 3) indicates the available binding pocket for CRP. The area of binding pocket is 24263.303Å2 and volume 53142.377Å3.
3.2. Retrieval of Ligands and ADMET Properties3.2.1. Ligand Chemical Structure. PubChem stands as the most extensive collection of openly accessible databases containing chemical information on a global scale. The ligands (Table 2) utilized in this study were selected from the most widely used database PubChem (https://pubchem.ncbi.nlm.nih.gov/).
Table 2. Ligand Retrieval from PubChem
|
S# |
Ligand Name |
Molecular Formula |
Molecular Weight |
3D Structure |
|---|---|---|---|---|
|
1. |
Apigenin |
C15H10O5 |
270.24 g/mol |
 |
|
2. |
Luteolin |
C15H10O6 |
286.24 g/mol |
 |
|
3. |
Orientin |
C21H20O11 |
448.4 g/mol |
 |
The selected phytochemicals of Rhapis excelsa include Apigenin, Luteolin, and Orientin identified as flavonoids.
3.2.2. Ligand Screening through Lipinski Rule and Toxicity. Lipinski's Rule of Five predicts the likelihood of a compound to be orally active if it has no more than 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors, a molecular weight under 500 daltons, and a log p-value not exceeding 5. These criteria help assess a compound's potential as an effective oral drug [15, 16]. According to the results generated by SwissADME (http://www.swissadme.ch/) for Lipinski rule of 5 the suitable ligands undergo further evaluation (Table 3).
Table 3. Applicability of Lipinski’s Rule of Five on Ligands
|
Ligands |
Lipinski rule violations |
|---|---|
|
Apigenin |
Yes; 0 violation |
|
Luteolin |
Yes; 0 violation |
|
Orientin |
No; 2 violations: NorO>10, NHorOH>5 |
The prediction of toxicities of compounds is a crucial component within the drug design and development process. Therefore, the ProTox-II tool (https://tox-new.charite.de/) was utilized for toxicity analysis.
Table 4. Toxicity Analysis
|
Toxicity Targets |
Predictions |
||
|---|---|---|---|
|
Apigenin |
Luteolin |
Orientin |
|
|
Hepatotoxicity |
Inactive |
Inactive |
Inactive |
|
Carcinogenicity |
Inactive |
Active |
Inactive |
|
Immunotoxicity |
Inactive |
Inactive |
Active |
|
Mutagenicity |
Inactive |
Active |
Active |
|
Cytotoxicity |
Inactive |
Inactive |
Inactive |
ProTox-II is an advanced web-based tool that is designed to predict the toxicity of chemicals using computational methods. It integrates several computational models, for instance, molecular similarity, pharmacophores, and machine learning, to assess various toxicity parameters, including acute toxicity, hepatotoxicity, carcinogenicity, and mutagenicity. The platform utilizes data from both laboratory assays and animal studies to generate accurate predictions. This functionality is highly beneficial for toxicologists, regulatory bodies, and pharmaceutical researchers, enabling them to evaluate the safety profiles of compounds effectively [15].
Based on the toxicity predictions provided by the ProTox-II (https://tox-new.charite.de/), it can be concluded that the flavonoid apigenin is quite suitable due to its demonstrated lack of activity against target toxicity (Table 4). In contrast, it has been observed that luteolin exhibits carcinogenic and mutagenic characteristics. Hence, apigenin, according to ADME properties and toxicity is identified as lead compound.
ADME parameters absorption, distribution, metabolism, and excretion describe the journey of drug through the body. Absorption refers to the drug entering the bloodstream, distribution refers to its spread through tissues, metabolism refers to its chemical transformation, and excretion refers to its removal from the body. These factors are essential to determine a drug's effectiveness and safety profile [17].
3.3. Glibenclamide and apigenin ADMET PropertiesThe structure of Glibenclamide was obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/).
Moreover, SwissADME (http://www.swissadme.ch/) was employed to acquire results pertaining to physicochemical properties and Lipinski rule. Glibenclamide and apigenin, SwissADME results were compared (Figure 4 and 5). Additionally, the toxicity evaluation was conducted using ProTox-II (https://tox-new.charite.de/). The results obtained represented no toxicity possibilities.
The molecular weight of apigenin was 270.24g/mol, whereas Glibenclamide was 494.00 g/mol. The Lipinski Rule showed 0 violations in apigenin and Glibenclamide. Apigenin showed high GI absorption, whereas Glibenclamide had low GI absorption. Apigenin seemed to exhibits excellent gastrointestinal absorption, while Glibenclamide demonstrateds comparatively low gastrointestinal absorption. Hence, Apigenin could be considered as a practical choice for a drug due to its superior gastrointestinal absorption characteristics. Based on these results, not much difference was observed between apigenin as a ligand and the drug Glibenclamide. Therefore, in the future, apigenin could be used as an anti-diabetic medication.
3.4. Molecular Docking between Standard Drug and Lead CompoundThe Glibenclamide structure retrieved from Pubchem (https://pubchem.ncbi.nlm.nih.gov/) was utilized for molecular docking through PatchDock (http://bioinfo3d.cs.tau.ac.il/PatchDock/). The highest docking score obtained was 6686. The visualization through UCSF Chimera (https://www.cgl.ucsf.edu/chimera/) of docking data was obtained by PatchDock (http://bioinfo3d.cs.tau.ac.il/PatchDock/).
UCSF Chimera is a comprehensive molecular visualization tool designed for interactive exploration and visualization as well as analysis of molecular structures and associated data. Created by the University of California, San Francisco, it
enables the visualization of atomic models, volumetric data, and sequence alignments. The tool includes features to generate high-quality images and animations, which aid in understanding the intricate details of biomolecular structures and their functions. Widely used in bioinformatics, structural biology, and medicinal chemistry, Chimera serves both educational and research needs [18].
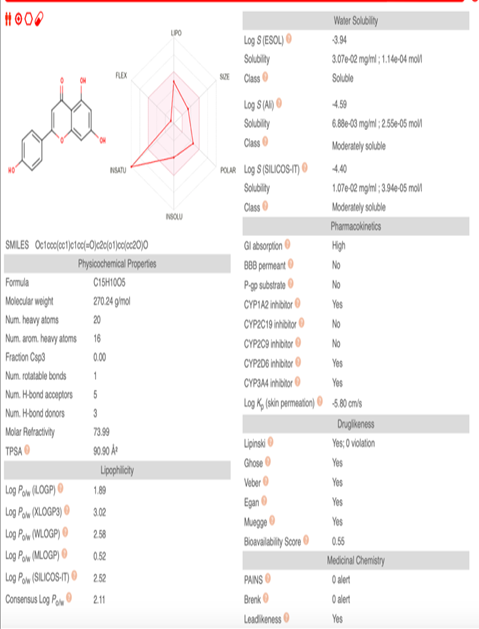
Figure 4. SwissADME Results of Glibenclamide
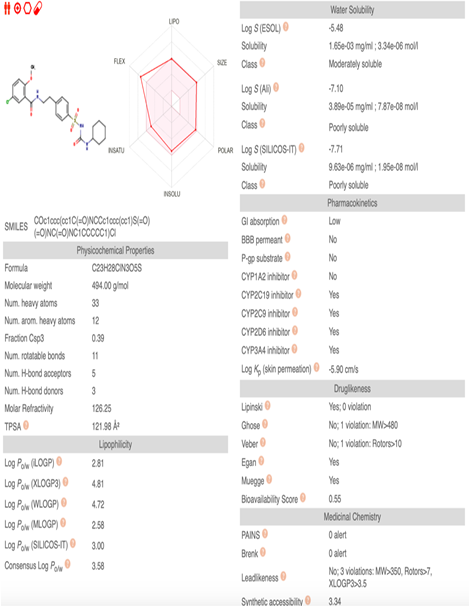
Figure 5. SwissADME Results of Apigenin
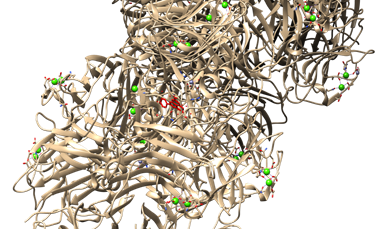
Figure 6. Protein-ligand Docking Visualization of CRP With Apigenin using UCSF Chimera. Red Color Indicates Docking Site
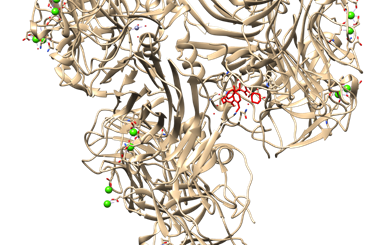
Figure 7. Protein-Ligand Docking Visualization of CRP with Glibenclamide using UCSF Chimera
In (Figure 6 and 7), the red colored region indicates the docking area. The lead compound apigenin exhibits binding affinity towards specific amino acids in the CRP.
4. DISCUSSION
In this study, computational methods including molecular docking as well as molecular dynamics simulations were used to investigate the possible anti-diabetic properties of Rhapis excelsa. The main goal was to investigate important phytochemicals that could work well with CRP, a biomarker linked to inflammatory reactions in diabetes.
The primary sequence of CRP was retrieved in FASTA format from the UniProt database (Accession number: P02741) to provide a solid basis for this research. Homology modeling, a crucial step in comprehending the three-dimensional (3D) structure of CRP, was performed using this sequence. To ensure the accuracy of the protein, the structure was verified using information from the Protein Data Bank (PDB). Additionally, the physicochemical properties of CRP were analyzed using ProtParam which yielded crucial information on the structural and functional features of the protein.
An important finding from the physicochemical evaluation was the GRAVY with a score of -0.052, suggesting that CRP is primarily hydrophilic. The analysis of physicochemical properties showed that the molecular weight of CRP is 24,925.42 Da with an isoelectric point (pI) of 5.45, indicating that it is slightly acidic at physiological pH. This acidity causes electrostatic interactions and solubility which ultimately affects the behavior of CRP in various biological as well as pathological conditions. The solubility of CRP and its ability to interact with water molecules and other biomolecules are significantly affected by its hydrophilic character. These molecular characteristics are especially important to analyze the functions of CRP as a biomarker in metabolic diseases and how it contributes to inflammation.
Phytochemicals from Rhapis excelsa were extracted from the PubChem database to find possible medicinal molecules. Flavonoids specifically, apigenin, luteolin, and orientin were preferred during the selection process due to their well-established bioactive properties. To determine the drug-likeliness, Lipinski's Rule of Five was used. Apigenin and luteolin satisfied all four parameters of Lipinski's. These compounds are suitable for drug development and have good oral bioavailability according to SwissADME analysis [16]. Orientin, on the other hand, has two exceptions of Lipinski's Rule that indicates low oral bioavailability and decreases its potential as a medicinal agent. Furthermore, apigenin was particularly shown to be a promising candidate in toxicity predictions that was done using ProTox-II. This is because it exhibited no detectable activity against toxicological targets. These results suggested that while orientin would need structural changes or different drug delivery methods to improve its effectiveness, apigenin and luteolin showed considerable promise as possible anti-diabetic substances.
PatchDock was used to carry out molecular docking which generated docking scores according to the binding affinity of each ligand to CRP. According to CASTp analysis, apigenin had the highest docking score of 4440 among the analyzed ligands. This suggested efficient and effective binding interactions with the active site of CRP [15]. However, it is important to interpret these computational results with caution, as no in vitro or in vivo validation was performed. Therefore, the therapeutic potential of apigenin remains a computational prediction at this stage and must be experimentally verified.
Glibenclamide, which is a common anti-diabetic drug, was also tested with CRP to validate these findings. Glibenclamide had a relatively higher docking score (6686) than apigenin, indicating a higher binding affinity [18]. However, in comparison of ADME (absorption, distribution, metabolism, and excretion) properties, there was a significant difference. Although, Glibenclamide and apigenin both followed Lipinski's Rule, Glibenclamide's absorption profile was lower than apigenin which showed higher gastrointestinal absorption. This indicates that apigenin is a suitable natural substitute for current anti-diabetic medications due to its potential for improved bioavailability and pharmacokinetic benefits [18]. This study contributed to the growing body of research. Furthermore, it explored the anti-diabetic effects of flavonoids and supporteds previous findings that link apigenin to improved glucose metabolism and anti-inflammatory effects [11, 12, 17]. By identifying CRP as a viable molecular target and confirming strong binding affinity of apigenin in silico, this study provided a foundation for further therapeutic exploration.
4.1. Conclusion
The current study identified potential phytochemical compounds from Rhapis excelsa using computational approaches for their application in diabetes drug development. Computational analyses including molecular docking and virtual screening revealed some bioactive compounds within Rhapis excelsa. This showed strong binding affinity to important molecular targets associated with diabetes. The identified phytochemicals exhibit characteristics that make them potential candidates for drug development in the field of diabetes management.
Among the screened compounds, apigenin demonstrated favorable docking scores and ADMET properties, suggesting its potential as a lead compound for further investigation. However, these findings are based solely on in silico methods and must be validated through experimental studies, including in vitro and in vivo assays. Therefore, while this study provided a valuable computational framework, additional biological and clinical research is essential before considering apigenin or any compound as a therapeutic agent.
Future work should also explore dynamic binding simulations, cellular toxicity assays, and comparison with multiple diabetic drug targets to support these computational predictions.
4.2. Limitations
Despite these promising computational outcomes, the study is limited by the absence of experimental data, lack of dynamic simulations, and testing against multiple standard drugs. Future study should include laboratory-based assays and animal models to validate the predicted efficacy and safety of apigenin.
Additionally, since CRP is associated with broader inflammatory responses, its targeting may have implications beyond diabetes potentially extending into cardiovascular and metabolic comorbidities, making this research relevant for long-term clinical exploration.
CONFLICT OF INTEREST
The authors of the manuscript have no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
DATA AVAILABILITY STATEMENT
The data associated with this study will be provided by the corresponding author upon request.
FUNDING DETAILS
No funding has been received for this research.
REFERENCES
- Jacob B, Narendhirakannan RT. Role of medicinal plants in the management of diabetes mellitus: a review. 3 Biotech. 2018;9(1):4. https://doi.org/10.1007/s13205-018-1528-0
- Adhikari B. Roles of alkaloids from medicinal plants in the management of diabetes mellitus. J Chem. 2021;2021:2691525. https://doi.org/10.1155/2021/2691525
- Han DG, Cho SS, Kwak JH, Yoon IS. Medicinal plants and phytochemicals for diabetes mellitus: pharmacokinetic characteristics and herb-drug interactions. J Pharm Investig. 2019;49(6):603–612. https://doi.org/10.1007/s40005-019-00440-4
- Singh P, Singh VK, Singh AK. Molecular docking analysis of candidate compounds derived from medicinal plants with type 2 diabetes mellitus targets. Bioinformation. 2019;15(3):179–188. https://doi.org/10.6026/97320630015179
- Kottaisamy CPD, Raj DS, Kumar VP, Sankaran U. Experimental animal models for diabetes and its related complications—a review. Lab Anim Res. 2021;37:23. https://doi.org/10.1186/s42826-021-00101-4
- Suryasa IW, Rodríguez-Gámez M, Koldoris T. Health and treatment of diabetes mellitus. Int J Health Sci. 2021;5(1):572192. https://doi.org/10.53730/ijhs.v5n1.2864
- Meo SA, Zia I, Bukhari IA, Arain SA. Type 2 diabetes mellitus in Pakistan: current prevalence and future forecast. J Pak Med Assoc. 2016;66(12):1637–1642. https://www.ncbi.nlm.nih.gov/pubmed/27924975
- Aamir AH, Ul-Haq Z, Mahar SA, Qureshi FM, Ahmad I, Jawa A, Sheikh A, Raza A, Fazal A, Jadoon Z, Ishtiaq O, Safdar N, Afridi A, Shehzad A. Diabetes prevalence survey of Pakistan (DPS-PAK): prevalence of type 2 diabetes mellitus and prediabetes using HbA1c: a population-based survey from Pakistan. BMJ Open. 2019;9(2):e025300. https://doi.org/10.1136/bmjopen-2018-025300
- Sangeetha R. Luteolin in the management of type 2 diabetes mellitus. Curr Res Nutr Food Sci. 2019;7(2):393–398. https://doi.org/10.12944/CRNFSJ.7.2.09
- Kanmani S, Kwon M, Shin MK, Kim MK. Association of C-reactive protein with risk of developing type 2 diabetes mellitus, and role of obesity and hypertension: a large population-based Korean cohort study. Sci Rep. 2019;9:4573. https://doi.org/10.1038/s41598-019-40987-8
- Miao L, Cheong MS, Zhou C, Farag M, Cheang WS, Xiao J. Apigenin alleviates diabetic endothelial dysfunction through activating AMPK/PI3K/Akt/eNOS and Nrf2/HO-1 signaling pathways. Food Front. 2022;4(1):420–431. https://doi.org/10.1002/fft2.192
- Vanaja D, Kavitha S. A study on phytochemicals, antioxidant activity and FT-IR analysis of Rhapis excelsa (Thunb.) A. Henry. Eur J Pharm Med Res. 2016;3(7):390–394. https://www.ejpmr.com/home/abstract_id/1128
- Biya A. The role of high sensitivity C-reactive protein level in predicting stroke and other cardiovascular events: a meta-analysis review. Int J Health Pharm. 2024;9(4):104–128. https://doi.org/10.56201/ijhpr.v9.no4.2024.pg104.128
- Stanimirovic J, Radovanovic J, Banjac K, Obradovic M, Essack M, Samardzija-Nenadov T, Jevtic A, Glisic S, Vojvodic N, Gojobori T, Isenovic ER. Role of C-reactive protein in diabetic inflammation. Mediators Inflamm. 2022;2022:3706508. https://doi.org/10.1155/2022/3706508
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263. https://doi.org/10.1093/nar/gky318
- Pollastri MP. Overview on the rule of five. Curr Protoc Pharmacol. 2010;49(1):9.12.1–9.12.8. https://doi.org/10.1002/0471141755.ph0912s49
- Anup N, Gadeval A, Rajpoot K, Tekade RK. Software used in ADME computation. In: Tekade AK, ed. Biopharmaceutics and pharmacokinetics considerations. London, UK: Elsevier; 2021:699–708. https://doi.org/10.1016/B978-0-12-814425-1.00006-1
- Surhone LM, Timpledon MT, Marseken SF. UCSF Chimera. Mauritius: Betascript Publishing; 2013. https://www.worldcat.org/title/ucsf-chimera/oclc/858876409