Zainab Asif Mirza, Ayman Naeem, Aamna Syed, Rana Muhammad Mateen, Muhammad Irfan Fareed, and Mureed Hussain*
Department of Life Sciences, School of Science, University of Management and Technology, Lahore, Pakistan
* Corresponding Author: [email protected]
Desbuquois dysplasia (DBQD) is an autosomal recessive chondrodysplasia that belongs to the multiple dislocation group and causes parental and afterbirth growth retardation, hand and proximal femur abnormalities, joint laxity, and scoliosis. Several missense and splice site mutations in CANT1 gene are linked with the development of DBQD. In silico approaches can predict the pathogenic variations causing hereditary diseases. Hence, in the current study, in silico analysis was used to forecast the variants of CANT1 gene that harm the functionality of calcium-dependent nucleotidase. A total of 281 variants with uncertain significance, retrieved from the gnomAD, dbSNP, ClinVar, and Variation Viewer databases, were analyzed using CADD, Meta SNP, CAPiCE, and Condel to predict 61 highly pathogenic variants. Stability change predicting computational tools were applied to filter 19 highly pathogenic amino acid variants that impact protein dynamics via sample conformation or during vibrational entropy. UCSF Chimera was used for interactive visualization and analysis of unwanted interaction among 5 variants in the molecular structure of the protein. Ligand binding computational tools were used to interpret the protein-ligand interactions. A total of three (3) post-translational modification sites were also predictably disrupted by 16 variants. Spice and HSF 3.1 tools were applied to 95 variants to check their disease-causing potential. The variants of the gene were analyzed using computational tools based on different algorithms. The most damaging variants of CANT1 gene that can affect the functionality and stability of the protein were predicted. It was determined that an extensive in silico analysis can determine the likely pathogenic variations for further in vitro experimental analysis.
Keywords: CANT1, Desbuquois dysplasia (DBQD), chondrodysplasia, gene mutations, gene variants, gnomAD
Genetics is a critical factor that plays a crucial role in all diseases [1, 2]. Many genetic mutations affect human beings in early stages of life and cause malformation of the body. According to skeletal disorder classification and nosology, more than 400 clinical phenotypes are divided into 42 groups [3]. Moreover, 1 out of 5000 children are born with skeletal disorders. Chondrodysplasia, short stature, deformed bones, patterning defects in digits, and joint dislocation are some of the skeletal abnormalities caused by genetic defects. Chondrodysplasia includes a number of diseases caused by changes in genes. Chondrodysplasia or skeletal dysplasia are rare diseases. If taken collectively, they comprise a group that generalizes skeletal affection and results in functional skeletal limitations and even mortality [4]. Desbuquois dysplasia (DBQD) is an autosomal recessive chondrodysplasia. Its characteristics include prenatal and after birth growth retardation, short arms and legs, joint laxity, and scoliosis [5]. Hand and proximal femur abnormalities are the main characteristics /of the patients with the disease. While short long bones with ‘Swedish key’ appearance are the main radiological features of this disease. ‘Swedish key’ is marked with exaggerated trochanter appearance and advanced hand and foot bone age with delta phalanx. Severe respiratory problems are also associated with DBQD. Cognitive impairment is the clinical feature that distinguishes DBQD from other dysplasia. On the basis of characteristic hand anomaly, two forms of DBQD have been identified. Beemer et al. [6] and Meinecke et al. [7] addressed the distinct skeletal dysplasia characterized by micro melic dwarfism, vertebral abnormalities, narrow chest, and advanced carpotarsal ossification. They considered it to be the same disorder that Desbuquois described in two sisters in 1992 and suggested that it is an autosomal recessive inherited disorder [8]. The pattern of autosomal recessive inheritance of DBQD is inveterate on the association of both parental genes, horizontal transmission, and cousin marriage reports in consanguineous families [9].
CANT1 and XYLT1 gene mutations have been recognized as causative genes. In 2009, calcium activated nucleotidase (CANT1) were suspected of causing DBQD. A member of apyrase family, CANT1 encodes soluble nucleotidase that favor the hydrolysis of UDP followed by GDP and UTP, although its function is still not specified in human beings. Nucleotidase has a very low hydrolysis activity towards ADP and ATP, however, the role of CANT1 in skeletal formation is still unknown [5].
CANT1 belongs to the apyrase family of enzymes comprising nucleoside di and triphosphate hydrolyzing enzymes. They play a role in maintaining hemostasis and cause the inhibition of platelet aggregation through the hydrolysis of adenosine diphosphate [10]. Apyrase enzymes also play a vital role in neurotransmission regulation [11]. A human soluble apyrase has been cloned and its activity rigorously depends on calcium. So, it has been named as human soluble calcium activated nucleotidase 1 (hSCAN-1). CANT1 encodes uridine diphosphate (UDP) nucleotidase, commonly required for the synthesis of proteoglycan. It is also involved in vesicular trafficking by calcium in Golgi apparatus through the activation of inositol triphosphate receptor [5]. It is responsible for DBQD and expands the hand anomaly spectrum in this disease [12]. It consists of 5 coding exons and three transcripts [13]. In 2009, Huber revealed that CANT1 variants caused DBQD Type 1 in 9 families closely followed by Huber and colleagues [14]. In 2011, Furuichi and others demonstrated that its variants were responsible for DBQD Type 2 [15]. CANT1 gene substrates are involved in many signaling pathways. After RT-PCR analysis, CANT1 gene showed specific expression in chondrocytes. This demonstrates that there is an electron dense material in distended rough endoplasmic reticulum in the fibroblasts of DBQD patients [5].
Homozygosity mapping technique has been used to study DBQD. That study recognized an infant with this disease whose parents were first cousins. The most important consequence of DBQD was mapped to 17q25.3. While combining the mapping results with others, this study narrowed the critical interval of the region that contained ten annotated genes. After the sequencing of these genes, it was revealed that a 5bp duplication in the CANT1 gene causes the disease. This report secured the role of CANT1 gene in DBQD [14]. In 2011, three families were reported with DBQD Type 1 in hydropic fetuses of gestational weeks 17, 22, and 25, respectively. X-ray studies indicated that there was delta-shaped extra phalangeal bones and disorder-specific eminence of lesser tubercle of the femur that varied with the age of fetus. The loss of function was associated with missense mutations when in vitro nucleotidase activity was measured [16]. In 2011, it was suggested that c.676G>A mutation of CANT1 gene in East Asian families should be derived from common families [17]. In 2015, three (3) Indian patients were reported with DBQD. These patients belonged to two different families. Previously, the mutation c.676G>A (p.Val226Met) in CANT1 gene was reported in DBQD patients from Japanese and Koreans. This study highlighted a novel homozygous mutation c.467C>T (p.Ser156Phe) and concluded that DBQD is not only present among East Asians but is also reported by the people of the Indian subcontinent [18]. Another report in 2019 revealed a Pakistani female patient diagnosed with DBQD Type 1 in three successive pregnancies. CANT1 gene was identified as the cause of the disease after comparing the regions of homozygosity within SNP microarray from the first two terminated fetuses. The parents were tested since confirmatory tests were prevented due to insufficient fetal DNA. Both parents had the heterozygous mutation of CANT1 ((c.643G>T; Glu215Term) gene. During the third pregnancy, ultrasound reports revealed a similar condition and the targeted analysis of the CANT1 gene recognized homozygous c.643G>T mutation. The study suggested that the utilization of SNP microarray identifies the list of candidate genes in those broods who are at risk of a rare autosomal recessive disorder [19].
Genetic mutations need to be identified because there is no cure for the diseases caused by these mutations. Pathways for these genetic mutations need to be predicted as well for better understanding of the diseases caused by this skeletal disorder. In computational bioinformatics, in silico approaches have gained much attention. These approaches use the computational tools and databases of different algorithms. Before going to wet lab, variations in genetic sequences, either deleterious, mutated, or neutral, are checked out in these computational approaches to save time and expense. Even the phenotypes of these mutated genotypes can be predicted [20].
To understand the mutation’s role in this gene, the current authors utilized computational tools [21] based on different algorithms [22], including missense tools to predict pathogenic variants, stability tools to analyze the impression of a mutated gene on the function of the protein, and post-translational modification analysis to detect the uncharacterized protein during translation from the genomic sequence [23].
Genetic difference is considered to be crucial in the study of human health and SNPs help predict the changes that may have adverse health effects. To study the effects of the SNPs of CANT1 gene, a total of 623 variants of this gene were retrieved from gnomAD (v2.1.1) [24], dbSNP, ClinVar, and Variation Viewer. After the application of filters on all the variants retrieved from different databases, 281 missense variants were obtained which were further analyzed for missense analysis.
Missense variants with allelic frequency ≤ 0.001 were then analyzed by CADD (https://cadd.gs.washington.edu) [25]. CADD scores the deleteriousness of single nucleotide variants. It scores variants according to their pathogenicity potential. CADD combines more than 63 annotations. CADD analysis is very important for further in silico analysis.. After CADD analysis, 157 missense variants with PHRED score higher than 20 were further analyzed by CAPICE (https://github.com/molgenis/capice), CONDEL (https://bbglab.irbbarcelona.org/fannsdb/) and Meta-SNP (https://snps.biofold.org/meta-snp/) computational tools that predict the highly pathogenic variants of the candidate gene.
After the missense analysis of variants and filter application, the remaining variants with their respective substitution of amino acid were further analyzed for stability check, using protein stability predicting tools. The stability of protein affects its structure , expression, and solubility. Furthermore, it also has an impact on its functions. Decreased stability in protein structure due to concerned mutation is indicated by a negative scoring value in stability analysis and vice versa is true in case of a positive scoring value. DynaMut (http://biosig.unimelb.edu.au/dynamut/) [26], DUET (http://biosig.unimelb.edu.au/duet/) [27], iStable 2.0 [28], and Foldx (http://foldxsuite.crg.eu/) plugin in YASARA (http://www.yasara.org/) [29] were used to analyze the impact of mutation on protein stability based on the change in Gibbs free energy .
Post-Translational Modifications (PTM) are modifications in which there attaches a specific group by covalent bond with the protein molecule, so that proteins can perform their function [30]. These modifications are usually enzymatic modifications of proteins or the particular protein process during or after its synthesis. To access the modification in protein that can be deleterious, the current authors used an online web-server ‘ScanProsite’ at ExPASy [31]. It determines the functions of uncharacterized protein during translation from its genomic sequence. It accurately identifies the family of protein to which the new sequence belongs and can also detect various types of modifications, such as phosphorylation, sumoylation, or palmitoylation. After the analysis of protein modifications, the structural analysis of protein was predicted using the tool NetSurf-3.0 (http://www.cbs.dtu.dk/services/NetSurfP/). This tool works on a neural network algorithm and predicts the secondary structure of amino acids in a sequence, as well as its structural disorder and backbone dihedral angles for each residue in the sequence. It also predicts the reliability of structures in the form of Z-score as ‘Buried’ or ‘Exposed’. Protein conservation analysis was carried out using the ConSurf (https://consurf.tau.ac.il/) server which is based on the evolutionary principle. Conserved and variable regions are indicated by the colors followed by MSA [32].
Wild and mutant type protein structures have clashes. These clashes can disturb the native conformation of a particular protein structure. In this study, UCSF Chimera (http://www.cgl.ucsf.edu/chimera/) [33] software was used for the visualization of clashes in CANT1 gene and energy minimization of these clashes. Structural energy minimization basically minimizes the molecules of the model, holding optically some atoms that are fixed. The Molecular Modeling Toolkit (MMTK) provides the minimization routines. Minimization is intended to clean small molecular structures and improve local interactions within the large molecule. Put simply, energy minimization moves the system toward a local minimum without crossing energy barriers and does not search for the global minimum. UCSF Chimera is designed on a clustering algorithm.
For splice site variants analysis, SPiCE [34], HSF 3.1 [35], and CADD were used. These softwares were used to access the pathogenicity on variations of CANT1 gene near splice sites. The aim was to evaluate the effects of mutations on splicing signals and to recognize splicing motifs in any human sequence.
A total of 281 variants of CANT1 gene were retrieved from these databases. Of these, 249 variants were retrieved after applying VEP annotation filter. Selected missense variants with allelic frequency ≤ 0.001 and allelic count < 50 were then analyzed using CADD. After applying the filter on variants with CADD scoring ≥ 20, 157 variants were left. CAPICE, CONDEL, and Meta-SNP were applied to measure the effect of single nucleotide polymorphism on pathogenicity and the SNPs associated with the disease were identified. Moreover, 95 variants (60%) were predicted to be deleterious out of the remaining 157 variants using CAPICE algorithm with the applied filter of ‘greater than or equals to 0.02’. Similarly, 133 variants (84%) were predicted to be disease associated by using CONDEL algorithm with the applied filter of ‘greater than 0.522’. Furthermore, 80 variants (51%) with R.I. value greater than 0.5 were predicted to be deleterious by using Meta-SNP algorithm. After the analysis, only 61 highly pathogenic missense variants were obtained.
In silico analysis was used to analyze Superoxide dismutase 3 (SOD3) in a study conducted in 2019. Molecular dynamic modeling revealed two mutations, namely p.A91T and p.R231G, to be detrimental for ligand binding study [36]. In 2009, Celine Huber along with others studied 9 DBQD families and identified 7 mutations in CANT1 gene. Among these were four nonsense mutations (Del 5′ UTR and exon 1, p.P245RfsX3, p.S303AfsX20, and p.W125X), while 3 mutations were missense mutations (p.R300C, p.R300H, and p.P299L). These mutations were identified as conserved amino acid changers at the seventh nucleotidase conserved region (NRC). In 5 out of 9 families, arginine substitution at position 300 was identified.
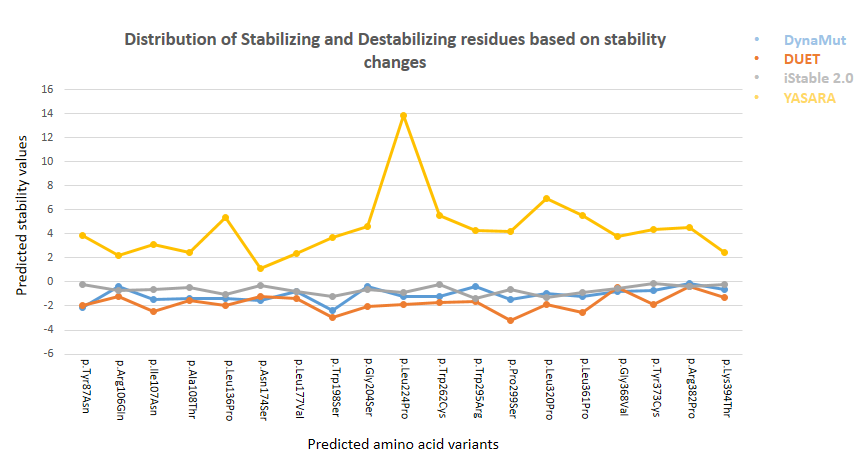
After missense analysis, 61 highly pathogenic missense variants were analyzed for stability check, using DynaMut, DUET, and iStable 2.0. The algorithm of all these stability tools defined negative ΔΔG as destabilizing, while positive values represented the stabilizing of the altered protein. DynaMut predicted 34 (55%) variants as destabilizing mutations which affected protein function and 27 (44%) variants as stabilizing mutations which were functionally troublesome. DUET predicted 55 (90%) variants as the most destabilizing variants and 6 (9.8%) variants as stabilizing variants. iStable 2.0 detected 11 variants (18%) as destabilizing mutations variants and 50 variants (81%) as stabilizing variants. The cut-off value applied on variants analyzed by stability tools was < 0 (for destabilizing variants). For cross-checking the stability of variants, YASARA was performed to predict protein stability. Alanine scan and stability analysis was done using YASARA, which indicated a positive value as destabilizing protein structure. Value binding free energy change (ΔΔG) for mutation were calculated in kcal/mol. FoldX determined the total energy of 61 variants in kcal/mol to predict the problem in protein structure. CANT1 gene variants stability checks due to amino acid substitution using stability tools are shown in Figure 1. Out of 61 variants, 19 were considered as highly functional and affected variants. In a recent study, missesnse variants in ligand binding pocket were characterized through SIFT, nsSNPAnalyzer, PolyPhen-2, PhD-SNP, SNPs&GO, and SNAP2. Stability analysis was performed by algorithm I-Mutant2 to predict the effect in Alzheimer’s disease [37].
Figure 1. Stability Analysis of Missense Variants
It was determined that 19 out of 61 variants destabilize the protein structure.
UCSF Chimera software was used to visualize clashes in CANT1 gene and for energy minimization of these clashes. CANT1 protein structure was retrieved from the RCSB PDB site. Clashes or unwanted contacts occur in the wild and mutant type structures of CANT1 protein. These clashes can disturb the native conformation of protein structure (Figure 4). No clashes were found in the wild type of Pro96Gln, Ser117Pro, Leu177Phe, Arg183Trp, and Lys394Arg. While, after structural energy minimization, there were clashes in the mutant types of these mutation residues, such as 1 clash in mutaPro96Gln, 1 in Ser117Pro, 1 in Leu177Phe, 4 clashes in Arg183Trp, and 1 clash in mutant types of Lys394Arg. Clashes in CANT1 gene due to mutations in protein structure using Chimera are shown in Figure 2.
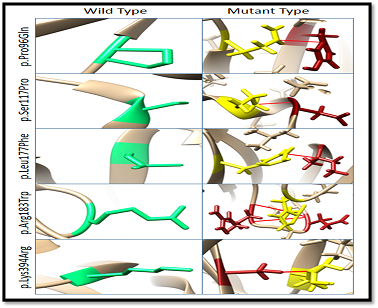
Figure 2. Clashes were Found in CANT1 Gene Due to Mutations in Protein Structure using Chimera
Clashes were observed in the mutant types of Pro96Gln, Ser117Pro, Leu177Phe, Arg183Trp, and Lys394Arg. Spring green colour indicates the wild type residue and fire brick color shows the mutant residue, whereas red lines show the contacts found and yellow indicates the interacting residues.
3.4. Post-Translational Modification (PTM) Analysis of CANT1 Gene
ScanProsite (EXPASY) was used to predict the post-translational modified sites in the structure of CANT1 (PDB. I.D 1S1D) and mutant sites were visualized by using UCSF Chimera. N-glycosylation site (Condition: -N-X-S/T, Here X=uncharged amino acid), protein casein kinase II phosphorylation site (Condition: -S/T-Aaa-Aaa-Aaa3-Aaa-Aaa, where Aaa shows acidic amino acids which must be present at +3 (Aaa3)), and protein kinase C phosphorylation site (Condition: -R/K1–3-X-S/T-X+1-(R/K)1–3, where X= uncharged amino acid and X+1=hydrophobic) were predicted as shown in figures 3a, 3b, and 3c.
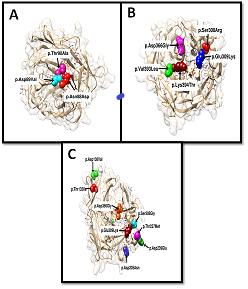
Figure 3. (A) Visualization of the loss of N-Glycosylation at positions p.Thr90Ala, p.Asp89Val, and p.Asn88Asp. These mutations disturb the motifs of NDTY. (B)Visualization of protein kinase C phosphorylation sites at positions p.Ser308Arg, p.Asp366Gly, p.Glu309Lys, P.Val393Leu, and p.Lys394Thr. Mutations p.Ser308Arg, p.Asp366Gly, and p.Glu309Lys disturb the motifs of SeK, while P.Val393Leu and p.Lys394Thr mutations disturb the motifs of SvK. (C) Visualization of protein casein kinase II phosphorylation sites at positions p.Asp138Val, p.Thr135Ile, p.Asp366Gly, P.Ser308Gly, p.Glu309Lys, p.Thr237Met, p.Asp239Glu and p.Asp205aAsn. Mutations p.Asp138Val, p.Thr135Ile affect the TIsD motifs, mutations p.Asp366Gly, P.Ser308Gly, p.Glu309Lys affect SekD motifs, mutations p.Thr237Met and p.Asp239Glu affect TtgD motifs, while p.Asp205aAsn affects the SdgD.
NetSurf-3.0 predicts the surface accessibility and secondary structure of amino acids in sequence, either as ‘Buried’ or ‘Exposed’. The results of conservational analysis using ConSurf and secondary structure analysis using NetSurf-3.0 data are shown below in Table 1.
Table 1. Results of Conservational Analysis using ConSurf and Secondary Structure Analysis using NetSurf-3.0
|
ConSurf |
NetSurfP-2.0 |
||||
|
Protein Change |
Score |
Grade |
RSA |
ASA |
Sec.Structure Exposed/Buried |
|
p.Asn88Asp |
9 |
-1.11 |
0.466 |
68.252 |
Exposed |
|
p.Asp89Val |
4 |
0.403 |
0.447 |
64.384 |
Exposed |
|
p.Thr90Ala |
8 |
-0.924 |
0.312 |
43.274 |
Exposed |
|
p.Thr135Ile |
3 |
0.558 |
0.249 |
34.509 |
Exposed |
|
p.Asp138Val |
1 |
2.162 |
0.658 |
94.847 |
Exposed |
|
p.Asp205Asn |
6 |
-0.306 |
0.566 |
81.604 |
Exposed |
|
p.Thr237Met |
2 |
0.751 |
0.579 |
80.238 |
Exposed |
|
p.Asp239Glu |
4 |
0.377 |
0.416 |
60.018 |
Exposed |
|
p.Asp366Gly |
3 |
0.462 |
0.522 |
75.206 |
Exposed |
|
p.Ser308Gly |
5 |
-0.091 |
0.504 |
59.069 |
Exposed |
|
p.Ser308Arg |
5 |
-0.091 |
0.504 |
59.069 |
Exposed |
|
p.Glu309Lys |
7 |
-0.643 |
0.58 |
101.378 |
Exposed |
|
p.Val393Leu |
1 |
1.265 |
0.31 |
47.708 |
Exposed |
|
p.Lys394Thr |
9 |
-1.184 |
0.419 |
86.229 |
Exposed |
ConSurf predicts conservational analysis in terms of color scale which represents the conservation scores (9 - conserved, 1 – variable). NetSurf-3.0 predicts surface accessibility in terms of RSA and ASA, as well as the secondary structure of amino acids in sequence, either as ‘Buried’ or ‘Exposed’.
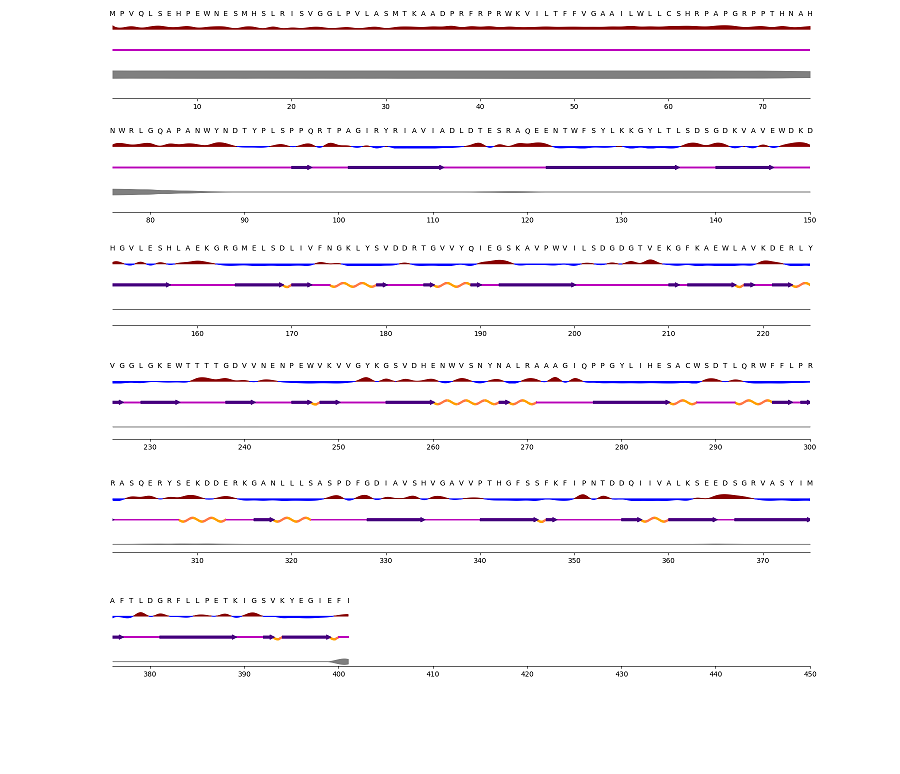
Figure 4. CANT1 structural analysis through NetSurf-3.0. Surface Accessibility: The red upward elevation indicates the exposed residue, while the sky blue low elevation shows the buried residue in protein structure. Secondary Structure: Straight pink line is the coil, the orange spiral is the helix, and the colored arrow (indigo) is the strand. Disorder: Below the secondary structure prediction line is the thick grayish line that shows the probability of disorder related to that residue. The wider the line is the greater are the chances of disordered residue.
A total of 184 variants were retrieved from GnomAD. Among these, 95 variants were considered as canonical splice site variants. These 95 variants were splice region variants. For spliceogenicity of the variants, they were tested by CADD. Afterward, the variants with a score greater than 15 were selected. The researchers were left with 10 variants which were further analyzed by splicing tools, namely SPiCE and Human Splice Finder 3.1 (HSF 3.1). Table 2 shows the results of splicing analysis. Fig 5 depicts the SPiCE graphical representation output of 10 variants based on SSF-like and MES scores alteration in percentage. Variants with blue area represent the probability of alteration under the decision threshold of optimal sensitivity and variants with red area represent the probability of the upper decision threshold of specificity.
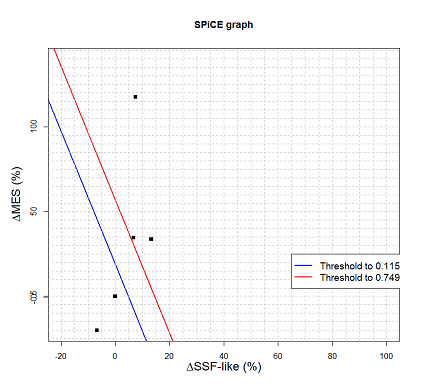
Figure 5. Graphical representation of SPiCE splicing tool of CANT1 gene. SPiCE graphical representation output of 10 variants based on SSF-like and MES scores alteration (in percentage). Variants with blue area represent the probability of alteration under the decision threshold of optimal sensitivity and variants with red area represent the probability of the upper decision threshold of specificity.
Table 2. Splice Site Variants’ Analysis in CANT1 Gene using Splicing Tools (CADD, SPiCE and HSF 3.1)
|
INPUT |
CADD |
Spice |
HSF |
||
|
Position |
PHRED |
Probability |
SPiCE Prediction |
HSF Prediction |
HSF Score |
|
17:78993923G>A |
15.99 |
0.96966 |
High |
No significant impact on splicing signals. |
No prediction |
|
17:78993925G>C |
10.8 |
0.02686 |
Low |
Broken W.T. Acceptor Site |
6.09>4.1 (-32.68%) |
|
17:78993926A>G |
6.929 |
0.80322 |
High |
No significant impact on splicing signals. |
No prediction |
|
17:78993929C>T |
5.206 |
0.00039 |
Low |
Broken W.T. Acceptor Site |
6.09>0.42 (-93.1%) |
|
17:78993944C>A |
5.63 |
0.02686 |
Outside SPiCE Interpretation |
No significant impact on splicing signals. |
No prediction |
|
17:78994946T>C |
5.141 |
0.02686 |
Outside SPiCE Interpretation |
No significant impact on splicing signals. |
No prediction |
|
17:78994992T>C |
5.065 |
0.02686 |
Outside SPiCE Interpretation |
New Acceptor splice site |
5.04>7.22 (43.25%) |
|
17:78995001G>A |
5.575 |
0.02686 |
Outside SPiCE Interpretation |
No significant impact on splicing signals. |
No prediction |
|
17:78995232A>G |
13.49 |
0.99979 |
High |
No significant impact on splicing signals. |
No prediction |
|
17:78996976C>A |
9.931 |
0.02686 |
Outside SPiCE Interpretation |
No significant impact on splicing signals. |
No prediction |
Desbuquois dysplasia (DBQD) is an autosomal recessive prenatal and postnatal growth disorder characterized by joint laxity, short extremities, and progressive scoliosis. Every 1 out of 5000 children are born with this skeletal disorder. In 2009, calcium activated nucleotidase (CANT1) was determined to be responsible for causing this disease. CANT1, a member of the apyrase family, encodes soluble nucleotidase that favour the hydrolysis of UDP followed by GDP and UTP, although its function remains unspecified in human beings. In this study, SNPs reported in CANT1 gene were analyzed to determine the cause of DBQD. Extensive in silico analysis helped the researchers to understand the genetics of this disorder and identify the mutations in CANT1 gene that cause this disorder. CADD, Meta SNP, CAPiCE, and Condel were used to examine 281 variants with unknown significance collected from the gnomAD, dbSNP, ClinVar, and Variation Viewer databases to forecast 61 highly harmful variants. The highly pathogenic 19 amino acid mutations were subjected to computational stability change prediction techniques. UCSF Chimera was utilized for the interactive display and investigation of undesirable interaction among five variants in the molecular structure of the protein. Moreover, 16 variants were projected to disrupt 3 PTM sites. Spice and HSF 3.1 tools were used to evaluate 95 variants for their propensity to cause disease and to forecast the ten splice variants with the highest pathogenicity. The reported SNP analysis of CANT1 gene can be further extended by following wet lab experimental work or it can be observed in animal models.