Humaira Amin, Asghar Shabbir, and Khuram Shahzad*
Department of Biosciences, COMSATS University Islamabad, Pakistan
* Corresponding Author: [email protected]
Alzheimer’s disease (AD) is an irreversible and progressive neurodegenerative disorder. The brain mechanisms involved in this disease remain largely unknown. Hence, this study used the integrated bioinformatics approach to analyze a high throughput sequencing dataset (GSE162873) in order to identify the potential biomarkers involved in the pathophysiology of this disease. DESeq2 package was used for the identification of differentially expressed genes (DEGs) from both healthy and diseased patients. DAVID, a web-based bioinformatics resource, was used to perform functional enrichment analysis. StringApp plugin in Cytoscape was utilized to construct the protein-protein interaction (PPI) networks, whereas hub genes were identified through cytoHubba. MCODE was used to perform module analysis, ClueGO to evaluate the KEGG pathways enriched in modules, and miRNet platform for the interaction analysis of miRNAs and hub genes. Drug-genes interaction analysis was performed using DGIdb resource to find out the related drugs. A total of 652 DEGs were screened which were significantly enriched in GO terms. KEGG pathways analysis showed that PI3K-Akt signaling, hippo signaling, MAPK signaling, TGF-beta signaling, and sphingolipid signaling were significantly enriched pathways. A total of 12 hub genes were found to be significantly interacting with miR-603, miR-10b-5p, miR-124-3p, and miR-1-3p, and some FDA approved drugs. The current study provided an insight into the molecular mechanisms of AD and identified some potential biomarker genes, their pathways, miRNAs, and drugs which might be useful for diagnostic and therapeutic purposes.
Keywords: Alzheimer’s disease (AD), hub genes, integrated bioinformatics, KEGG pathways, RNA-sequencing, transcriptome analysis.
Alzheimer’s disease (AD) is a serious neurological disorder which is progressive in nature. It causes the necrosis and shrinkage of brain cells which leads to gradual memory loss, difficulty in thinking, and other cognitive deteriorations to such an extent that a person cannot function independently [1]. WHO categorizes AD under dementia. The worldwide prevalence of this disease was assessed to be 50 million, with 10 million new cases annually [2] . The most common risk factor for AD is ageing and increasing age is directly linked with the disease [3]. Other common associated risk factors include obesity, hypertension, type 2 diabetes, pre-existing cerebrovascular diseases, depression, head injury, impaired hearing, physical inactivity, and smoking [4–6] .
Alzheimer is marked by extracellular accumulation of the plaques of Amyloid-β (Aβ) and the formation of intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated tau-protein, resulting in abnormalities of limbic and cortical areas of brain. These plaques accumulate initially in the temporal, basal, and orbitofrontal neocortex areas and then slowly spread throughout the hippocampus, amygdala, neocortex, basal ganglia, and diencephalon. Thus, NFTs and Aβs are the major players associated with the progression of the disease [7, 8] . This disease can also be familial or sporadic [9] . There are two types of ADs namely early-onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD). The former starts to develop before the age of 65 years and the latter shows its symptoms after the age of 65 years. Among these, EOAD is initiated by mutations in presenilin genes (PSEN1 and PSEN2) and the Aβ precursor protein (APP) gene [10] . Whereas, the initiation of LOAD is associated with various types of gene-environment interactions. These interactions may include different gene polymorphisms that cause metabolic alterations such as hyperinsulinemia, altered adipokines, and hypercholesterolemia. Other possible causes of this type of AD include head injury, hypertension, psychological stress, and aluminum toxicity [11] . In LOAD, Aβ slowly accumulates in the brain before the appearance of the symptoms [12] . Among the three common variants (E2, E3, and E4) of apolipoprotein E (APOE), the primary genetic risk factor associated with LOAD is the E4 allele [13] . Several potential biomarkers and therapies for the identification and cure of AD have been identified; however, these remedies only temporarily slow down the progression of the disease. There is no effective treatment strategy available which can stop disease progression in the brain [14] .
The identification of key molecules which may be involved in causing cognitive impairment at an early stage has been very challenging. Several studies have established that early-stage neuro impairment is strongly impacted by the changes in genes regulation and expression. These findings suggest that the understanding of transcriptome may help to improve the diagnostic strategies for AD [15, 16]. With the recent advancements in sequencing technologies, the involvement of bioinformatics analysis for the identification of the biomarkers of diseases has rapidly increased.
In the current study, several biomarkers of AD were identified using the publicly available transcriptomics dataset available at the Gene Expression Omnibus (GEO) database [17]. Different bioinformatics techniques were utilized to analyze and perform the enrichment analysis of the KEGG (Kyoto Encyclopedia of Genes and Genome) pathways and gene ontology (GO) terms. Moreover, the functional association of genes was determined through the construction of protein-protein interaction (PPI) networks, which were used further to identify the hub genes and modules. The interaction of the retrieved hub genes with miRNAs and FDA approved drugs was also underscored.
2.1. Retrieval and Analysis of RNA Sequencing Data
RNA-sequencing dataset with identifier code GSE162873 [18] was retrieved using the GEO database, a public repository for microarrays, RNA-sequencing, DNA-sequencing, and gene ChIP expression datasets. This dataset was obtained through GPL11154 Illumina HiSeq 2000 (Homo sapiens) platform on AD. The dataset consisted of 8 tissue samples (4 samples from AD patients and 4 samples from healthy individuals). The raw counts of each sample were used for the subsequent differential expression analysis.
2.2. Data Normalization and Processing
The normalization and processing of count matrix containing raw counts from each sample was performed through DESeq2 package. DESeq2 utilizes the Wald test for calculating probability values, Benjamini Hochberg (BH) method for the correction of the values, and the maximum likelihood estimate (MLE) for gene expression [19]. Further, the adjusted p-value < 0.001 and |log2FC| >1 criteria were followed to screen differentially expressed genes (DEGs). The expression of DEGs was visualized through heatmap which was created using pheatmap package [20] available in R Bioconductor. The volcano plot of the acquired DEGs was also constructed to show the overall differential expression trend.
2.3. Enrichment Analysis of Selected Genes
Functional enrichment analysis was executed to explore DEGs. For this purpose, DAVID (Database for Annotation, Visualization, and Integrated Discovery) bioinformatics resource [21] was used to obtain the gene ontology (GO) terms and KEGG (Kyoto Encyclopedia of Genes and Genome) pathways. The cutoff criteria for the functional analysis were p-value <0.05 and gene counts > 4.
2.4. Network Construction and Extraction of Hub Genes and Modules
The functional and physical interactions of DEGs were assessed using stringAPP plug-in in Cytoscape, which uses STRING database to provide information about both functional associations and physical interactions of the genes [22]. The threshold confidence score for the generation of network was ≥ 0.4. These networks were visualized using Cytoscape [23]. The top 20 genes were identified using 5 different topological methods, namely degree, density of maximum neighborhood component (DMNC), edge percolated component (EPC), maximum clique centrality (MCC), and maximum neighborhood component (MNC). These genes were mined from the PPI network through the application of cytoHubba. It provides a user friendly interface to extract hub objects and nodes from biological networks [24]. The genes found by using at least 4 different methods were considered hub genes. MCODE, which identifies clusters within a network [25], was used to extract modules from the PPI network using default parameters. The modules are densely connected regions in a PPI network representing a group of proteins involved in performing specific functions [26]. The cutoff criteria used for the screening of modules were score > 5 and nodes > 5. The screened modules were then subjected to KEGG pathways enrichment analysis in Cytoscape using ClueGO plugin which generates dynamic network structures of these pathways enriched with the genes of interest [27]. The pathways having p-value < 0.05 were selected for analysis.
2.5. miRNA-mRNA Interaction Network Analysis
Nervous tissue specific miRNAs and hub genes interaction was predicted through a web-based tool namely miRNet 2.0. It covers 11 databases to provide high quality data on miRNA-target interaction [28]. Nervous tissue specific miRNAs were chosen because AD primarily affects the nervous system [29]. Cytoscape was used to visualize the resulting co-interaction networks.
2.6. Drug-Gene Interaction Analysis
The interaction of hub genes with potential drugs was also evaluated. For this purpose, an online database DGIdb (Drug-Gene Interaction database) v4.2.0 was used. This database uses web-based sources as well as the text mining of publications and different databases to provide interaction information of drugs with respective genes [30].
3.1. Identification of Significant Genes
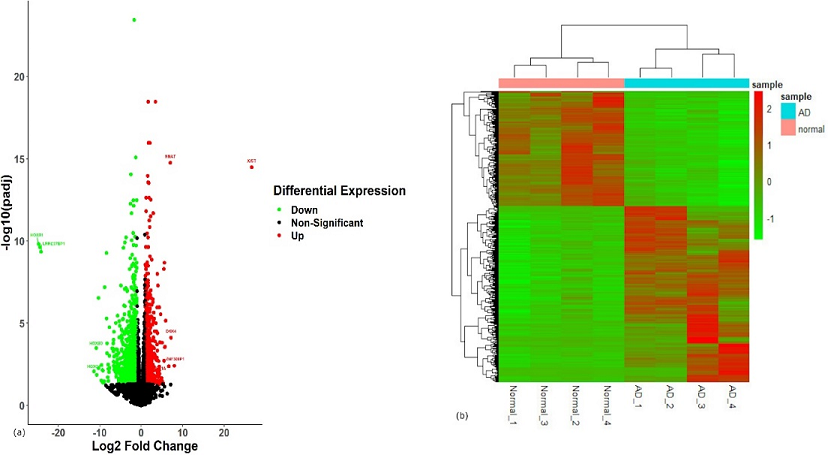
Following the cutoff criteria, 652 DEGs were short listed from the total genes list. Out of these, 394 genes were up-regulated, and 258 genes were down-regulated. The visualization of DEGs is represented through volcano plot and heatmap (Figure 1 (a) and Figure 1(b)).
Figure 1. (a) Volcano Plot of Differentially Expressed Genes (DEGs). The Upregulated Genes are Shown in Red Color, While the Downregulated Genes are Shown in Green. Black Color Represents the Insignificant Genes. (b) Heatmap of DEGs. The Upregulated Genes are Shown in Red Color, While the Downregulated Genes Are Shown in Green
3.2. Enrichment Analysis of Selected Genes
Functional enrichment analysis of the selected genes revealed that 627 genes were enriched in both GO terms and KEGG pathways. Among the GO terms enrichment results, a total of 10 biological processes (BP) were found to be enriched. Among these BPs, the top 5 included protein kinase B signaling, angiogenesis, anterior/posterior pattern specification, negative regulation of apoptotic process, and ureteric bud development. The top 5 GO terms from the cellular components (CC) category included axon, neuronal cell body, dendrite, lysosomal lumen, and sarcolemma, while the molecular functions (MFs) found to be significantly enriched included metal ion binding, actin binding, growth factor activity, microtubule binding, and sequence specific DNA binding. Dot plot of the GO terms plotted through ggplot2 package [31] in R software (v4.0.2) is shown in Figure 2. This dot plot displays the names of the GO terms (BP, CC, MF) along with the gene counts and associated p-values.
3.3. Networks, Hub Genes, and Modules
PPI network consisting of 360 nodes and 778 interactions is shown in Figure 4(a). cytoHubba discovered 100 hub genes using the 5 methods mentioned above, out of which 12 hub genes were found to be common across 4 different methods namely MNC, Degree, MCC, and EPC, as shown in Figure 4(b). These 12 hub genes were TIPM1, SPARC, P4HB, GAS6, FST, COL6A1, GPC3, TGFB1, ITGA5, COL6A2, COL1A2, and CYR61 (Table 1).
Under the specified threshold of K-score > 5 and nodes > 5, 2 modules were screened from the PPI network with K-score 8.4 and 5.125 and nodes 16 and 17, respectively (Figure 4(c) and Figure 4(d)).
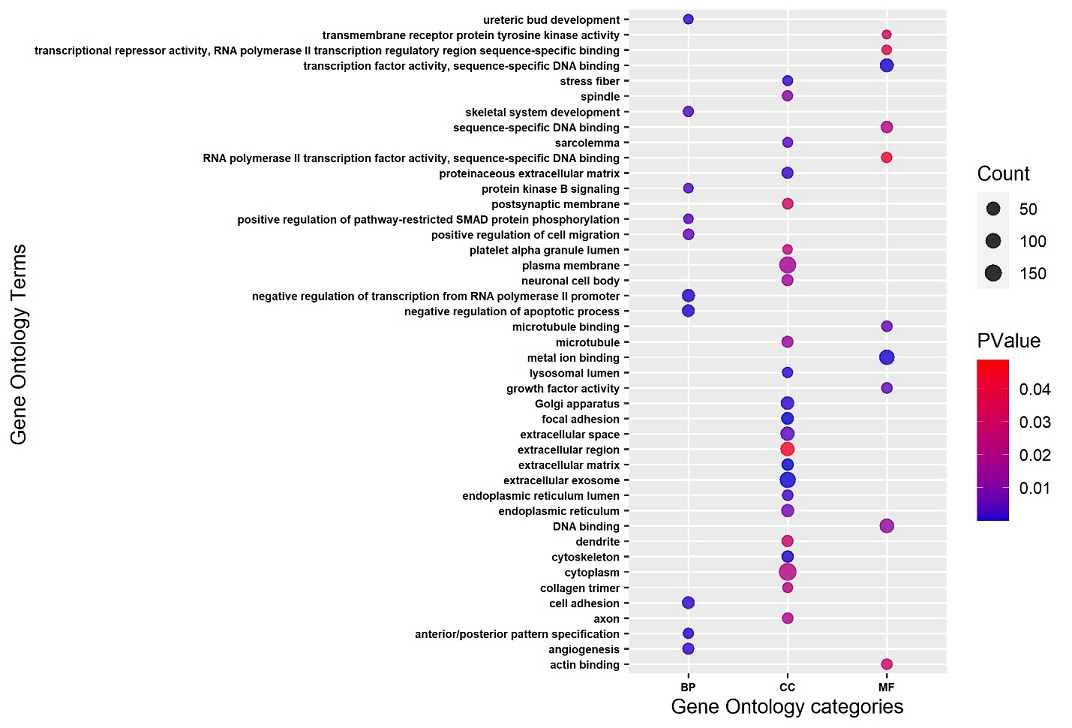
Figure 2. The GO Terms (BP, CC, and MF) Enriched with DEGs from AD are Shown in the Dot Plot. The Size of the Dots Depicts the Number of Genes Involved in Each GO Term, While Dot Color Represents the Corresponding p-Value
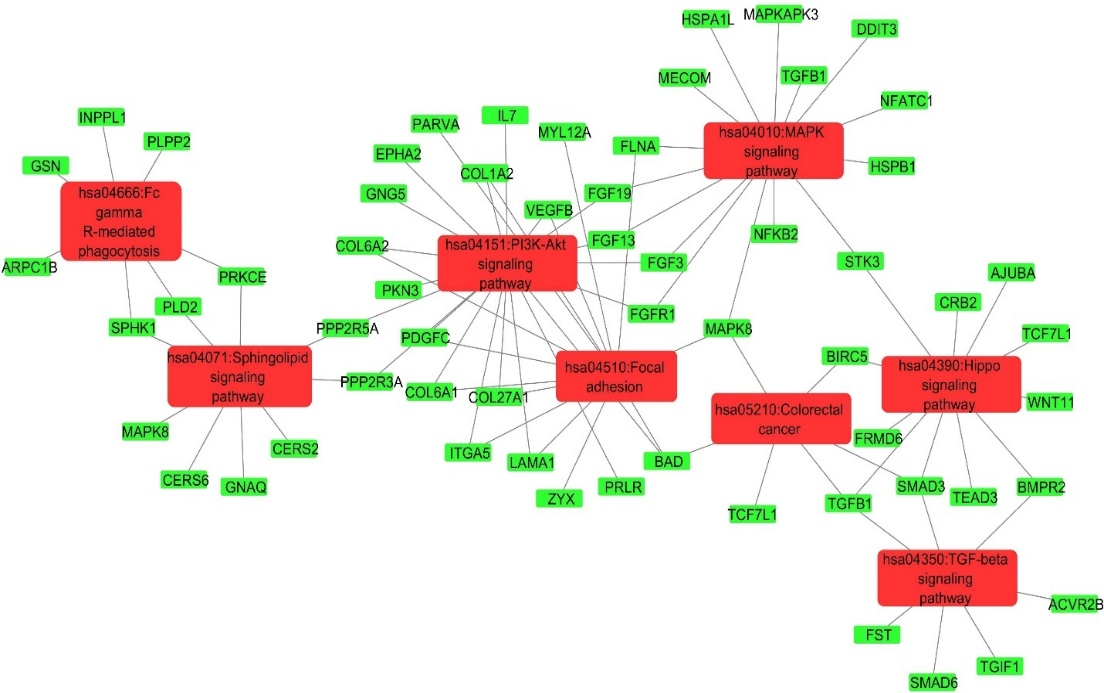
Figure 3. KEGG Pathways Enriched with DEGs from AD Patients. Red Boxes Display KEGG Pathways’ IDs and Names, While Green Boxes Represent DEGs.
Table 1. Gene Symbols, Log Transformed Expression Values, and Adjusted p-Values Of 12 Hub Genes Found Via 4 Different Methods of cytoHubba (MNC, Degree, MCC, and EPC)
|
Sr. # |
Gene symbols |
log2FC |
Adjusted p value |
|
1 |
GAS6 |
1.312106 |
8.35E-16 |
|
2 |
COL1A2 |
3.94514 |
1.21E-10 |
|
3 |
P4HB |
1.099993 |
2.06E-09 |
|
4 |
COL6A2 |
2.439654 |
0.000000991 |
|
5 |
FST |
2.096529 |
0.00000728 |
|
6 |
COL6A1 |
1.591406 |
0.0000307 |
|
7 |
TIMP1 |
1.947849 |
0.000123516 |
|
8 |
ITGA5 |
2.747075 |
0.000225242 |
|
9 |
SPARC |
1.181922 |
0.000242704 |
|
10 |
TGFB1 |
1.235326 |
0.000327422 |
|
11 |
CYR61 |
2.279453 |
0.000368363 |
|
12 |
GPC3 |
1.881975 |
0.000789719 |
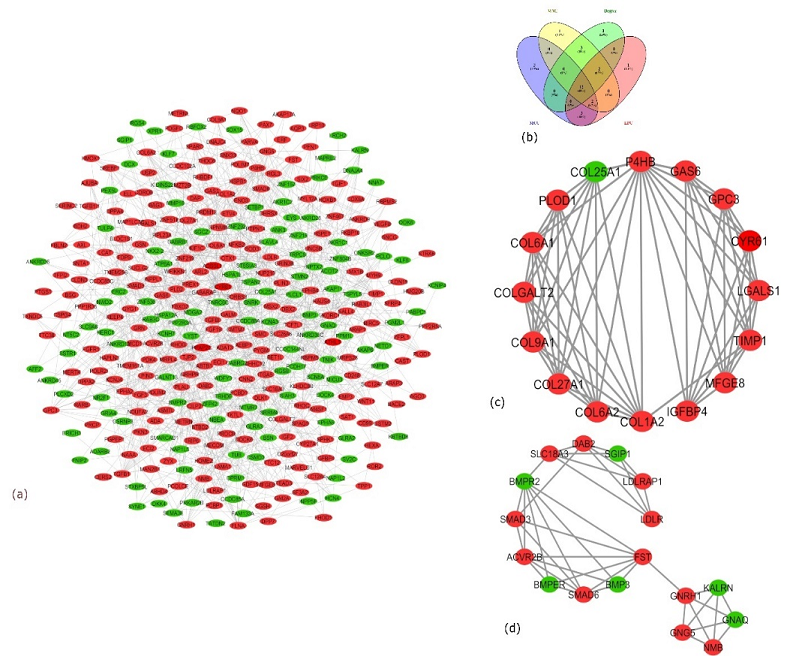
Figure. 4 Protein-Protein Interaction (PPI) Networks, Hub Genes, and Their Modules. (a) PPI Network of DEGs from AD. Red Nodes Display the Upregulation of Genes, While Green Nodes Represent the Downregulation of Genes. (b) Venn Diagram Represents the Number of Common Genes Found Via 4 Different Methods of cytoHubba. The Four Methods Used Were Degree, Maximum Neighborhood Component (MNC), Maximum Clique Centrality (MCC), and Edge Percolated Component (EPC). (c) Module 1 was Extracted from the PPI Network with the Score 8.4 and 16 Nodes. (d) Module 2 was Extracted from the PPI Network with the Score 5.125 and 17 Nodes
3.4. KEGG Pathways Enrichment of Modules
The enrichment of the KEGG pathways of the modules extracted from the PPI network showed that a significant number of KEGG pathways were enriched with DEGs. These included TGF-beta signaling, protein digestion and absorption, cholesterol metabolism pathways, and ECM receptor interaction (Figure 5(a) and Figure 5(b)).
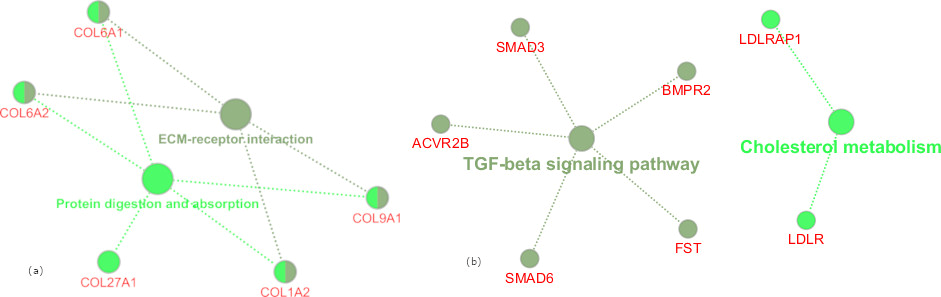
Figure 5. KEGG Pathways Enrichment of Modules (a) KEGG Pathways Enriched in Module 1, (b) KEGG Pathways Enriched in Module 2.
3.5. miRNA-mRNA Interaction Network
The co-interaction network of the 10 hub genes with 9 miRNAs was identified. The network consisted of 26 edges and 19 nodes which were visualized through Cytoscape and is shown in Figure 6. Two hub genes (TIMP1 and GPC3) were excluded as no miRNA was found interacting with them. ITG5 (degree = 4), P4HB (degree = 4), and TGFB1 (degree = 3) were the top 3 hub genes with the most miRNA interactions.
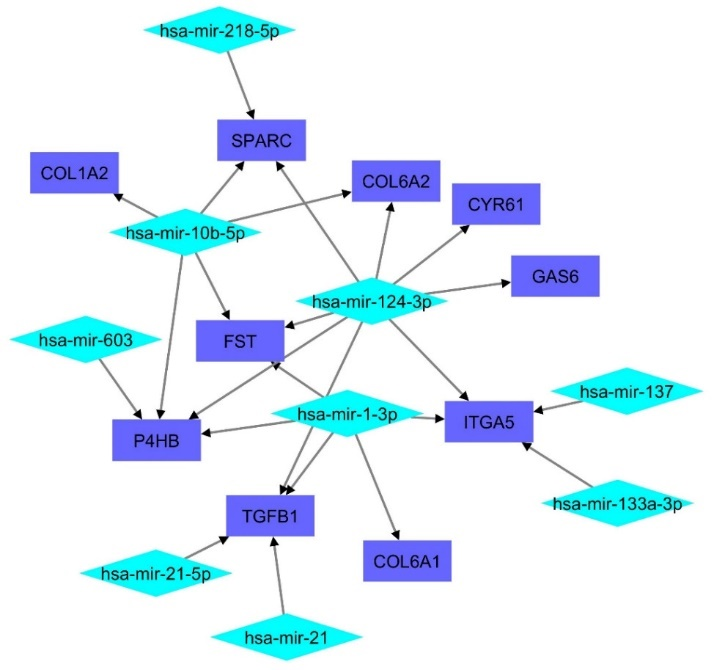
Figure 6. MicroRNA-hub Genes Interaction Network. The Interaction Between the miRNAs and Hub Genes is Represented Through Arrows. The Rectangular Nodes Represent the Hub Genes, and the Diamond Shaped Nodes Represent miRNAs.
3.6. Drug-Gene Interaction Analysis
FDA approved drugs were retrieved from the DGIdb resource. A total of 75 drugs were found to interact with hub genes. The potential drug targets were TGFB1, P4HB, and ITGA5 because most of the drugs were found to interact with these 3 hub genes. There was no drug information available against two hub genes namely CYR61 and GPC3. Drug-gene interaction network visualized through Cytoscape is shown in Figure 7.
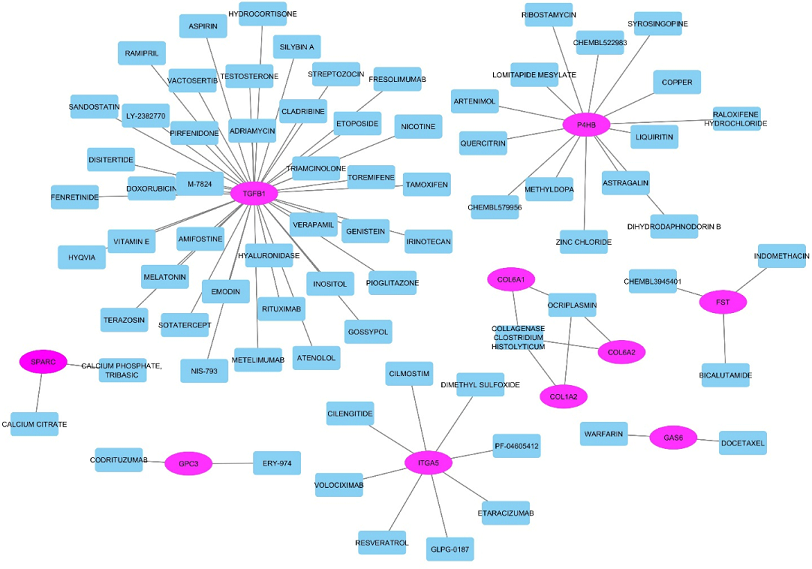
Figure 7. Drug-gene interactions network. Oval shaped nodes represent gene names, while rectangular nodes represent the interacting drugs. The lines represent the interaction between genes and drugs.
Despite the advancement in the field of clinical management, there is no treatment strategy available which can cure AD. The study of gene expression using RNA-sequencing technology is a promising technique to achieve a deeper insight into the possible molecular mechanisms of the disease [32].
In the current study, integrated bioinformatics approach was implemented to find out the potential biomarkers involved in AD. A significant number of DEGs were screened from the analyzed dataset.
GO terms enrichment analysis of DEGs identified Protein Kinase B (PKB) signaling, angiogenesis, and negative regulation of apoptotic process as significant biological processes. The activation of PKB signaling is involved in the progression of AD because the increased level of PKB corresponds to the increased level of hyperphosphorylated tau protein involved in neurofibrillary degeneration [33]. Negative regulation of apoptotic processes is associated with therapeutic effects on the progression of the disease [34]. If this process is disrupted, the disease progresses. Activated angiogenesis in neurons results in the accumulation of Aβ and hence is linked with cognitive decline in AD [35]. Among the molecular functions, metal ion binding, actin binding, growth factor activity, microtubule binding, and sequence specific DNA binding were found to be significant. For normal brain function, the hemostasis of metal ion needs to be established. Both the deficiency and excessive deposition of metal ions including Zn, Cu, and Mg are correlated with AD. The reason is that they may either induce oxidative stress or stimulate the overproduction and accumulation of Aβ [36]. Actin binding function of the proteins also has functional importance. For example, Cofilin-1 protein is an actin binding protein and a major regulator for actin cytoskeleton (CSK). It is involved in the development of AD because when it is phosphorylated it causes synaptotoxicity and synaptic impairment [37]. The activity of growth factors, especially the nerve growth factors (NGFs), is important in the progression of AD. When the metabolism of NGFs is altered it becomes involved in an imbalance between the NGF precursor mediated pro apoptotic signaling and TrkA mediated survival pathway which causes cholinotrophic neuronal dysfunction [38]. Microtubule associated proteins (MAPs) like tau have clear implications in AD. A characteristic of the pathological condition is the detachment of tau from microtubules and the formation of intracellular inclusions through its aggregation as paired helical filaments (PHFs) [39]. In neurodegenerative diseases, the accumulation of hyperphosphorylated and conformationally changed tau has been proved by different studies [40]. Hence, the molecular functions related to microtubule are important in AD.
The KEGG pathways enrichment analysis exhibited that DEGs were primarily enriched in PI3K-Akt signaling, hippo signaling, MAPK signaling, TGF-beta signaling, and sphingolipid signaling pathways. The inhibited PI3K-Akt signaling pathway is linked with the progression of AD. Aβ oligomers inhibit this pathway, thus inhibiting the activities of PDK1 and RTKs. It leads to the increased activity of GSK-3β which results in excessive tau phosphorylation and consequently induces neurodegenerative disorders [41]. This pathway can be treated as a potential target in the treatment of AD. Activated MAPK signaling pathway was found to be involved in AD because it induces tau hyperphosphorylation, thus preventing synaptic plasticity and inducing neural apoptosis [42]. Impaired TGFβ signaling results in decreased clearance of extracellular Aβ aggregations and increased neuroinflammation, leading to neurodegeneration [43]. Aberrant Fc-gamma receptor-mediated phagocytosis was also found to be involved in the pathophysiology of AD [44].
A total of 12 hub genes were found in the dataset, as shown in Table 1. In the current study, these hub genes were found to be involved in different pathways. These pathways mainly included PI3K-Akt signaling, focal adhesion, Hippo signaling, MAPK signaling, ECM-receptor interaction, TGF-beta signaling, and regulation of actin cytoskeleton. Among these hub genes, a higher expression of TIMP1 gene was observed in the cerebrospinal fluid of the patients with neurodegenerative disorders [45]. The second hub gene SPARC was also colocalized with the deposition of Aβ protein. Its upregulation is likely involved in innate immune response triggered by the neuropathology of AD [46]. It has been established that the upregulation of SPARC is linked with destructive effects on blood-brain barrier (BBB) which worsens the AD condition [47]. As far as another hub gene GAS6 is concerned, a recent study [48] confirmed the association of its upregulation with the progression of AD, although the mechanism was not described. The upregulation of COL6A1 and COL6A2 genes from the collagen VI family has been detected in the brain samples of AD patients. Their upregulation is associated with neuroprotective effects in AD. The possible mechanism for this effect is the ability of these genes to block the interaction between neurons and Aβ oligomers [49]. The role of TGFB1 has been established in developing late-onset AD (LOAD). Several studies have suggested that single nucleotide polymorphisms (SNPs) in neurotrophic factors such as TGFB1 and BDNF pose an increased risk of developing LOAD [50, 51]. It has been demonstrated that the overexpression of ITGA5 may indicate the reduced risk of dementia [52, 53].
The analysis of miRNA and hub genes revealed that there may be a strong association of miRNAs with hub genes in the pathogenesis of AD. Figure 6 shows that 9 miRNAs were found to interact with 10 hub genes. Among these 9 miRNAs, miR-603 has a protective effect in AD [54], while miR-10b-5p has an antiapoptotic effect. Hence, it may also have neuroprotective effect [55]. Another miRNA namely miR-124-3p reverses the neurodegenerative process in the brain by inhibiting the process of tau protein’s abnormal hyperphosphorylation when it targets Caveolin-1 and regulates Coveolin-PI3K/Akt/GSK3β. MiR-1-3p, which was found to interact with 5 hub genes, acts as a direct regulator of an anti-apoptotic molecule known as Fas apoptotic inhibitory molecule (FAIM). This microRNA regulated protein may act as a therapeutic target against neurodegenerative disorders [56]. MiR-137 negatively correlates with the expression levels of SPTLC1 and SPTLC2 which results in increased production of Aβ in AD [57]. Dysregulated miR-21-5p is related with the pathology of Aβ in AD [58]. Hence, the interacting microRNAs were determined to have a significant role in neurodegenerative disorders. Drug-gene interaction analysis of hub genes using DGIdb retrieved 77 FDA approved drugs which showed a strong relation with hub genes (see Figure 7). TGFB1 gene was found to interact with 40 drugs which is the highest number among the identified interactions. The figure shows that P4HB and ITGA5 were found to interact with a significant number of drugs (with 14 and 8 drugs, respectively).
4.1. Conclusion
The current research helps to improve our understanding about the progression of AD. The currently identified hub genes and pathways may be targeted as biomarkers for diagnostic and treatment purposes. It was also found that nervous-tissue specific miRNAs have a significant role in AD and may be used as therapeutic agents. FDA approved drugs that interact with the hub genes may be explored further to find new treatments against the disease. Taken together, the integrated bioinformatics approach has provided several GO terms, pathways, hub genes, miRNAs, and drugs related to AD which may lead towards new strategies to treat this disease.
4.2. Limitations
The current study has some limitations as well since the dataset consisted of only 8 samples. Hence, these findings need to be tested on a broader scale as well as through in vitro and in vivo studies to check their validity.