
| Review | Open Access |
|---|
In silico Evaluation of the Inhibitory Potential of Novel Hybrid Efflux Pump Inhibitors againstMycobacterium tuberculosis |
 |
|---|
![]() Ali Raza1*, Akmal Ali2, Siddiqa Batool1, Sheikha Rehman3, and Kazim Raza4
Ali Raza1*, Akmal Ali2, Siddiqa Batool1, Sheikha Rehman3, and Kazim Raza4
1Human Resource Development Center (HRDC), Health Department, Skardu, Pakistan
2Department of Biological Sciences, University of Baltistan, Skardu, Pakistan
3Department of Physiotherapy, RHQ Hospital, Skardu, Pakistan
4Department of Oncology & Research Center, RHQ Hospital, Skardu, Pakistan
Tuberculosis (TB) is an infectious condition caused by Mycobacterium tuberculosis. One of the key steps towards introducing infectious disease in control is the creation and application of antibiotics. M. tuberculosis strain is multidrug resistant, which is a major threat to TB control. It develops multidrug resistance (MDR) by using efflux pumps (EPs) and other associated systems that can reduce the efficacy of the drug. Several techniques are currently developed to overcome the efflux-mediated resistance and the development of efflux pump inhibitors (EPIs) is one of them. The current study aims to provide an in-silico evaluation of biologically validated hybrid efflux pump inhibitors (HEPIs) with different M. tuberculosis EP proteins. Twelve different HEPIs were identified through literature review. Docking analysis was used to examine the role of HEPI inhibition against 5 MDR EPs. Additionally, the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of all hybrid EP inhibitors were assessed. Molecular docking indicated that several HEPIs, specifically 5, 7, and 11, showed persistent higher binding affinities across multiple proteins, with docking scores comparable to or better than already known inhibitors. The predicted ADMET profiles suggested that most inhibitors had good oral bioavailability and adequate safety margins. To conclude, these HEPIs have the ability to effectively inhibit TB in human beings. In this regard, HEPI-5, HEPI-7, and HEPI-11 were determined as the most promising inhibitors because of their high binding affinities and positive ADMET profiles. Although experimental validation is essential to confirm their therapeutic relevance, these findings highlight their potential as novel EPI scaffolds. However, some of their pharmacological properties are not appropriate for human beings.
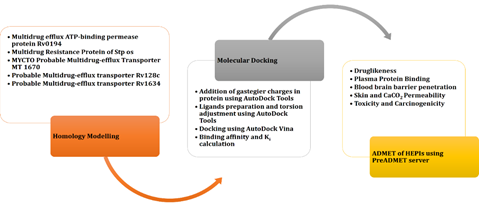
1. INTRODUCTION
Tuberculosis (TB) is a contagious infection caused by Mycobacterium tuberculosis. TB is considered a competing disease with human immunodeficiency virus (HIV) and a major public health hazard due to the number of deaths it causes worldwide [1]. The spread of the disease is exacerbated by poor economic conditions and 95% of patients suffering from this disease are from developing or underdeveloped countries [2]. Financial problems, food quality and insecurity, illiteracy, poor housing, and environmental conditions are the major causes of TB in developing countries. The World Health Organization (WHO) reported that approximately 10 million people are infected with TB worldwide. In Pakistan, about 510,000 TB cases are reported annually. Approximately 15000 developing drug-resistant TB cases are also reported annually. Globally, Pakistan ranks the 4th highest in terms of the prevalence of multidrug-resistant TB [3]. The main reasons for the occurrence of drug-resistant TB are inappropriate and inadequate drug regimens, unsupervised treatment, poor follow-up, lack of social support program for high-risk groups, and delay in diagnosis. High-risk groups for TB disease include people with HIV infection, homeless people, refugees, prisoners, and alcohol users [4, 5].
The discovery and use of antibiotics are among the most significant actions towards the prevention of infectious diseases. Impacts of pathogenic organisms due to their acquired antibiotic resistance have made most of the available antibiotics ineffective [6]. Extensive knowledge regarding the molecular mechanism of microbial antibiotic resistance is needed to cope with the rising number of drug-resistant bacteria and multidrug resistance (MDR) bacteria that possess different modes to protect against the harmful action of drugs [7]. MDR strains of tuberculosis poses a significant risk to the TB control process. These strains evolve through the action of M. tuberculosis via the efflux pumps (EPs) and other such comparable mechanisms that have the ability to limit the actions of drugs [8]. Originally, it was found that the first antibiotic efflux was released in 1980 when the chain of resistance against tetracycline was attributed to enterobacteria [9]. The EPs of M. tuberculosis perform functions that are unrelated to medication. Some of them are substrate specific [10-12] and, therefore, have a role in the assembly of MDR forms. In selective release of particular antibiotics, the EPs of bacteria, in contrast to other MDR pumps, release certain structurally varied compounds [13, 14].
Various tactics are aimed at offsetting efflux-mediated resistance and the development of efflux pump inhibitors (EPIs) is one of them. They can be prepared by (i) enriching the structural design of already known antibiotics or (ii) via the use of novel compounds to reduce or eliminate pump activity [15]. The counteracting drug efflux is still a relatively untried field in TB drug discovery and a good attacking method to reduce serious obstacles in the treatment of TB. Various molecular mechanisms have been observed to add efficacy to anti-TB drugs, in vivo, in vitro, and in the macrophage. These substances consist of verapamil (VER), phenothiazines, thioridazine, and chlorpromazine [16]. There is a dire need to design and synthesize EPIs for TB. As reported earlier, different hybrids improve the activities of rifampin (RIF) and isoniazid (INH) against extracellular, as well as intracellular M. tuberculosis. The hybrids include verapamil and thioridazine substructures, as well as the analogue of thioridazine [17]. Additionally, these agents might be used to decrease the probability of resistance against newly discovered drugs and to resuscitate the previously out of control anti-TB agents that were discarded owing to EP-mediated drug resistance [18].
The main liability of EPIs is their administration in combination with antibiotics which poses an extra challenge from a pharmacological perspective. In case of TB, major toxic pharmacokinetic interactions between rifampin and verapamil have limited additional studies of this combination. To overcome this possible liability, structural hybrid efflux pump inhibitors (HEPIs) are worth exploring. These ensure the delivery of pharmacologically optimized EPIs to the disease site. HEPIs are designated to be practiced in combination with an antibiotic like RIF. To prevent deleterious pharmacokinetic interaction, there still remains the need to optimize pharmacokinetic parameters. HEPIs are designed through the fusion of verapamil substructure with different non-tricyclic and tricyclic cores of chemosensitizers or their structural motifs derived through diphenylmethane, dibenzosubery, dibenzazepine, thioxanthene, phenothiazines, and cyproheptadine [19, 20]. All these chemosensitizers are known to reverse anti-mycobacterial drug resistance and to sensitize the resistant strains of M. tuberculosis by using different types of machinery, such as EPI. There is the possibility that these chemosensitizers do not exhibit intrinsic anti-mycobacterial activity. Various HEPIs have been derived that depict improved features of drug efflux inhibition which could be optimized further [18].
The current study involves the in silico analysis of experimentally–tested, biologically verified, and ethically approved HEPIs with various EP proteins of M. tuberculosis. By way of literature, 12 hybrid EPIs were determined [21]. This was done to perform the docking analysis of the role of inhibition played by HEPIs with reference to 5 MDR efflux proteins. Moreover, through the PreADMET the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of all hybrid EPIs were determined.
2. MATERIALS AND METHODS
The current study targets different multidrug EPs. There is no crystal structure available for these proteins. Therefore, homology modeling was performed to model their tertiary structure. HHpred was used to find the homologous proteins of EP proteins and to perform the homology modeling [22] (Table 1). On the basis of sequences, best templates were selected and model validation was performed using PROCHECK [23] and ProSA-web [24]. A total of 12 hybrid EPIs were discovered through literature [21]. Their chemical names and structures are given below in Table 2.
Table 1. PDB IDs of Template Proteins used for Different Proteins.
|
S. No. |
Protein Name |
Template Protein’s PDB ID |
|---|---|---|
|
1 |
Multidrug efflux ATP-binding permease protein Rv0194 |
5KO2_B, 4F4C_A 6BHU_A 5UJA_A 6BAA_F |
|
2 |
Multidrug Resistance Protein of Stpos |
4LDS_B 1PW4_A 4GC0_A 4ZP0_A 5OXO_A |
|
3 |
MYCTO Probable Multidrug-efflux Transporter MT 1670 |
4ZP0_A 1PW4_A 4LDS_B 3O7Q_A 5OXO_A |
|
4 |
Probable Multidrug-efflux transporter Rv128c |
4LDS_B 5AYN_A 2CFQ_A 1PW4_A 4ZP0_A |
|
5 |
Probable Multidrug-efflux transporter Rv1634 |
4ZP0_A 1PW4_A 4LDS_B 3O7Q_A 5OXO_A |
Table 2. Chemical Name and Structure of Compounds.
|
Compound Name |
Chemical Name |
Structure |
|---|---|---|
|
Comp-1 |
5-((2-(10H-phenothiazin-10-yl)ethyl)(methyl)amino)-2-(3,4-dimethoxyphenyl)-2- Isopropylpentanenitrile (1a) |
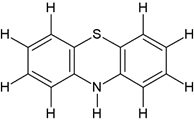 |
|
Comp-2 |
5-((2-(2-chloro-10H-phenothiazin-10-yl)ethyl)(methyl)amino)-2-(3,4-dimethoxyphenyl)-2- isopropylpentanenitrile (1b) |
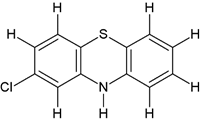 |
|
Comp-3 |
5-{[2-(10,11-Dihydro-dibenzo[b,f]azepin-5-yl)-ethyl]-methyl-amino}-2-(3,4-dimethoxyphenyl)- 2-isopropyl-pentanenitrile 1(c) |
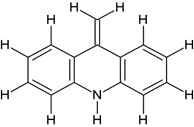 |
|
Comp-4 |
5-((2-(4-(10H-phenothiazin-10-yl)piperidin-1-yl)ethyl)(methyl)amino)-2-(3,4- dimethoxyphenyl)-2-isopropylpentanenitrile (2a) |
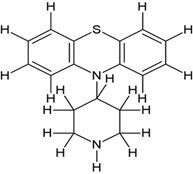 |
|
Comp-5 |
5-((2-(4-(2-chloro-10H-phenothiazin-10-yl)piperidin-1-yl)ethyl)(methyl)amino)-2-(3,4- dimethoxyphenyl)-2-isopropylpentanenitrile (2b) |
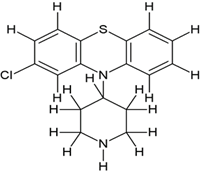 |
|
Comp-6 |
5-((2-(4-(5H-dibenzo[a,d][7]annulen-5-ylidene)piperidin-1-yl)ethyl)(methyl)amino)-2-(3,4- dimethoxyphenyl)-2-isopropylpentanenitrile (3a) |
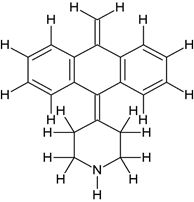 |
|
Comp-7 |
5-((2-(4-(9H-thioxanthen-9-ylidene)piperidin-1-yl)ethyl)(methyl)amino)-2-(3,4- dimethoxyphenyl)-2-isopropylpentanenitrile (3b) |
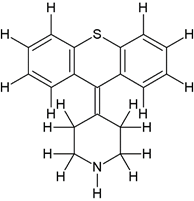 |
|
Comp-8 |
5-((2-(4-(2-chloro-9H-thioxanthen-9-ylidene)piperidin-1-yl)ethyl)(methyl)amino)-2-(3,4- dimethoxyphenyl)-2-isopropylpentanenitrile (3c) |
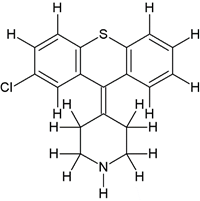 |
|
Comp-9 |
5-({2-[4-(10,11-Dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-ethyl}-methylamino)- 2-(3,4-dimethoxy-phenyl)-2-isopropyl-pentanenitrile (4). |
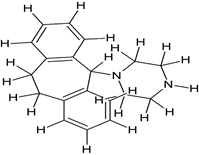 |
|
Comp-10 |
5-{[2-(Benzhydryl-amino)-ethyl]-methyl-amino}-2-(3,4-dimethoxy-phenyl)-2-isopropylpentanenitrile (5a). |
 |
|
Comp-11 |
5-((2-(benzhydryloxy)ethyl)(methyl)amino)-2-(3,4-dimethoxyphenyl)-2- isopropylpentanenitrile (5b) |
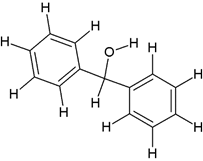 |
|
Comp-12 |
5-{[2-(4-Benzhydryl-piperazin-1-yl)-ethyl]-methyl-amino}-2-(3,4-dimethoxy-phenyl)-2- isopropyl-pentanenitrile (6). |
 |
Docking analysis was used to examine the inhibition of hybrid EPIs as compared to 5 MDR EPs. It prepared the ligands and proteins using AutoDock tools and calculated docking using AutoDockVina [25, 26]. ACDChemSketch optimized the 3D structure of HEPIs in energy and minimized the energy of the structures [27]. In all the receptors and ligands (HEPIs), polar hydrogen was added by using AutoDock tools. Polar charges helped to enhance the interaction processes. The ligands also demand access to patient torsions, so torsion modification was introduced in the ligands. This demonstrates the kinds of torsions required to treat and accomplish docking. The 3D grid was devised to define the search space in receptors. RaptorX was used to determine the binding pockets [28]. The interaction of the ligands (HEPIs) and the estimation of binding energies were analyzed with the help of Autodock Vina.
2.2 ADMET and Prediction of Drug LikenessBy using PreADMET, the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of all HEPIs were calculated [29]. The program also aided in predicting the drug-likeness of the given HEPIs. These predictions were prepared by using the file of their molecular structure. Using the pharmaceutically relevant properties associated with the ligand molecules and significant descriptors, the pharmacokinetics and pharmacological properties of the HEPIs were analyzed.
3. RESULTS
The protein model of the EPs was not available. The homology model validation showed that the EPs showed good stereochemical and structural quality. ProSA-web Z-scores for all proteins fell within the range of experimentally determined structures of a similar size (between –7.5 and –10.2). A total of 5 different EPs showed similarity with different proteins. At the primary structural level, the multidrug efflux ATP-binding permease protein Rv0194 showed maximum similarity with the MDR protein of Mus musculus and Caenorhabditis elegans, bovine MDR protein of Bostaurus, and ATP-sensitive inward rectifier potassium channel of Rattusnorvegicus.
MDR protein Stp showed maximum similarity with bicyclomycin resistance protein of Staphylococcus epidermidis, Glycerol-3-phosphate transporter, D-xylose-proton symporter, and multidrug transporter of Escherichia coli. MDR protein also showed similarity with Di-or tripeptide:H+ symporter, as well as Alpha-helical membrane protein of Streptococcus thermophiles.
MYCTO probable multidrug-efflux transporter MT1670 showed maximum similarity with the multidrug transporter of Escherichia coli (strain K12), Glycerol-3-phosphate transporter and L-fucose-proton symporter of Escherichia coli, and bicyclomycin resistance protein of Staphylococcus epidermidis and Di-or tripeptide:H+ symporter of Streptococcus thermophilus.
The probabilcy multidrug-elefux carrier Rv1258c was comparable with bicyclomycin resistance protein of Staphylococcus epidermidis, solute carrier family 39(Iron-regulated; alpha-helcthe; transporter protein) of Bdellovibriobacteriovorus lactose permease, and multidrug transporter of Escherichia coli.
Rv1634, which is likely the transporter of multiple drugs, was similar to multidrug transporter of Escherichia coli (strain K12), bicyclomycin resistance protein of Staphylococcus epidermidis, G + L-fucose:H+ pronucleid of Escherichia coli, and Di-or tripeptide:H + protein of Streptococcus thermophiles.
The HEPIs were all docked to various EPs in order to determine the binding energy and the inhibitory constant (Ki). Due to docking, it was discovered that all HEPIs exhibited varied behaviour when interacting with various EP proteins. Tables 3-7 exhibit the binding affinities (ranging between -3.0kcal/mol and -10.8kcal/mol).
3.1. Docking with Multidrug Efflux ATP-binding Permease Protein Rv0194Comp-1 / Comp-2 HEPIs were docked at the binding pockets of multidrug efflux ATP-binding permease proteins Rv0194, Arg952, and Pro957. Comp-2 and Comp-1 binding affinities were -5.2 and -5.5 kcal/mol, respectively. The other two HEPIs, namely Comp-3 and Comp-8, docked the binding affinities of -5.5 and -6.9 kcal/mol, respectively. The remnants of such interactions were Ala958 and Tyr951. The binding affinity of -5.0 kcal/mol was found to dock Comp-4. The outcome of the interaction included the residues of Leu576 and Val578; His152. Comp-5 docked to Val578, His1152, and His1170 with a binding affinity of -5.2, -6.9, and -7.5 kcal/mol, respectively. These interactions were with GLY979 and Tyr951. These residues had binding affinities of -3.0 and -5.1 kcal/mol, respectively. The binding of Comp-11 and Comp-12 was also seen with p952, p994, and p950 and the binding affinities were -5.2 and -4.9 kcal/mol, respectively. These interactions were with the two residues of A954 and T951 (Table 3).
Table 3. Binding Affinities of All Compounds with Multidrug Efflux ATP-binding Permease Protein Rv0194.
|
Compound Name |
Interactions Diagram |
Binding Affinity (kcal/mol) |
Ki (µM) |
|---|---|---|---|
|
Comp-1 |
 |
-5.2 |
152.807 |
|
Comp-2 |
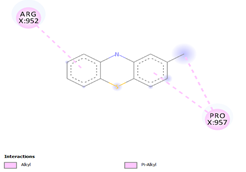 |
-5.5 |
92.044 |
|
Comp-3 |
 |
-5.5 |
92.044 |
|
Comp-4 |
 |
-5.0 |
214.243 |
|
Comp-5 |
 |
-5.2 |
152.807 |
|
Comp-6 |
 |
-7.5 |
3.136 |
|
Comp-7 |
 |
-6.9 |
8.643 |
|
Comp-8 |
 |
-6.9 |
8.643 |
|
Comp-9 |
 |
-3.0 |
6288.452 |
|
Comp-10 |
 |
-5.1 |
180.936 |
|
Comp-11 |
 |
-5.2 |
152.807 |
|
Comp-12 |
 |
-4.9 |
253.681 |
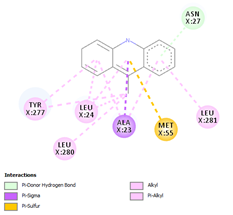
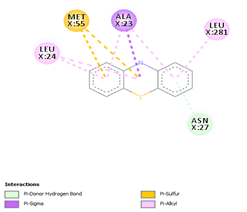
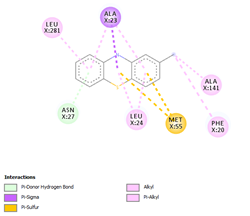
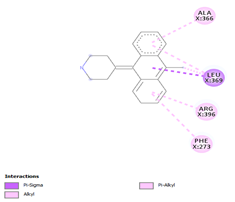
Comp-1 ligand was designed to dock at the binding pocket of the MDR protein of Stp. Comp-1 had the binding affinity of -6.7 kcal/mol. The residues observed by Met55 during the interaction were Ala23, Asn27, Leu24, and Leu281. The strong binding affinity of Comp-2 was measured at 7.0 kcal/mol but the interacting residues were Ala23, Leu 24, Asn27, Ala 141, Phe 20, Met 55, and Leu 281. The binding affinity of Comp-3 was 7.5 and the binding factors were Asn27, Tyr 277, Leu24, Ala23, Leu280, Met55, and Leu281. Comp-4 was found to be docked with the binding affinity of -6.6 kcal/mol. The binding affinity of Comp-5 was -6.9 kcal/mol and Comp-5 interacted with the residues Ala23, Asn27, Leu24, Met55, Phe20, and Leu281. Comp-6 bound to the MDR protein of Stp at A366, L369, A396, and P273 with a high binding affinity of -9.6 kcal/mol. Comp-7 and Comp-8 showed the binding affinity of 8.6kcal/mol. The residues that interacted with Comp-7 included Ala141, Phe 273, Arg 396, Leu369, and Ala 366. The associated residues involved in the interaction of Comp-8 were Ala366, Phe273, and Arg396, as well as Ala141. The weakest binding specificity attached to Comp-9 was -3.8 kcal/mol and the one-residue interaction site was Asn27. Comp-10 had a binding specificity of -7.4 kcal/mol and the interaction resolution sites released Leu24, Leu281, and Tyr277. Comp-11 and Comp-12 exhibited identical binding affinities. They had an affinity against each other of -7.3kcal/mol. Phe404, Leu 281, Ser 400, Tyr 277, Met 55, Ala 23, Leu 24, and Arg 396 were the residues involved in the interaction of Comp-11. Comp-12 interaction residues were Leu44, Leu282, Leu407, and Leu281 (Table 4).
Table 4. Binding Affinities of All Compounds with MDR Protein of Stpos
|
Compound Name |
Interaction Diagram |
Binding Affinity (kcal/mol) |
Ki (µM) |
|
|---|---|---|---|---|
|
Comp-1 |
 |
-6.7 |
12.118 |
|
|
Comp-2 |
 |
-7.0 |
7.299 |
|
|
Comp-3 |
 |
-7.5 |
3.136 |
|
|
Comp-4 |
 |
-6.6 |
14.348 |
|
|
Comp-5 |
 |
-6.9 |
8.643 |
|
|
Comp-6 |
 |
-9.6 |
0.090 |
|
|
Comp-7 |
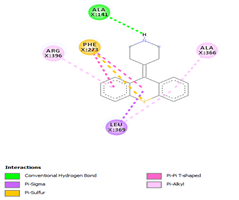 |
-8.6 |
0.489 |
|
|
Comp-8 |
 |
-8.6 |
0.489 |
|
|
Comp-9 |
 |
-3.8 |
1627.379 |
|
|
Comp-10 |
 |
-7.4 |
3.713 |
|
|
Comp-11 |
 |
-7.3 |
4.397 |
|
|
Comp-12 |
 |
-7.3 |
4.397 |
|
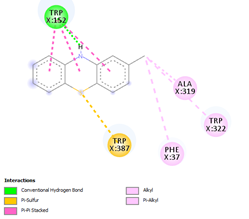
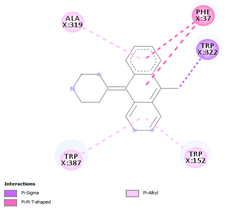
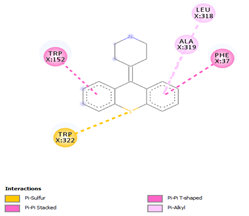
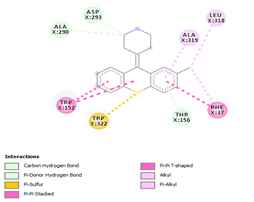
Comp-1 docked at the binding pocket of MYCTO probable multidrug-efflux transporter MT1670 with Phe37, Ile383, Ala290, and Trp387. The binding affinity of Comp-1 was -7.2 kcal/mol. Comp-2 docked with the binding affinity of -7.4 kcal/mol. The residues involved in the interaction were Phe37, Ala290, Ile383, Leu318, and Trp387. The binding affinity of Comp-3 was -8.1 kcal/mol. Whereas, Phe37, Trp387, Ile383, and Ala290 were the residues involved in the interaction. Further, Ile383, Ala290, Phe37, and Trp387 were the residues involved in the interaction for Comp-4 and the binding affinity was -7.2 kcal/mol. Com-5 was docked with the binding affinity of -7.6 kcal/mol and the residues involved in the interaction were Trp152, Ala319, Trp322, Phe37, and Trp387. Comp-6 showed the maximum binding affinity with this protein which was -10.8 kcal/mol. The residues involved in the interaction were Ala319, Phe37, Trp322, Trp152, and Trp387. Comp-7 and Comp-8 had the same binding affinities. Their binding affinity was -10.0 kcal/mol. The residues involved in the interaction for Comp-7 were Leu318, Ala319, Phe37, Trp152, and Trp322. While, the residues found in the interaction for Comp-8 were Ala290, Asp293, Ala319, Leu318, Phe37, Thr156, Trp322, and Trp152. Comp-9 showed the binding affinity of -3.9 kcal/mol and the residues involved in the interaction were Leu124 and Trp87. The residues involved in the interaction for Comp-10, Comp-11, and Comp-12 were Trp152, Ala125, Leu124, and Val29, respectively. Comp-10 and Comp-12 had the binding affinities of -8.1 kcal/mol and -8.4 kcal/mol, respectively (Table 5).
Table 5. Binding Affinities of All Compounds with MYCTO Probable Multidrug-efflux Transporter MT 1670.
|
Compound Name |
Interaction Diagram |
Binding Affinity (kcal/mol) |
Ki (µM) |
|---|---|---|---|
|
Comp-1 |
 |
-7.2 |
5.206 |
|
Comp-2 |
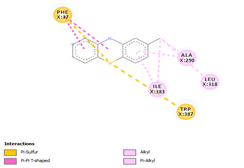 |
-7.4 |
3.713 |
|
Comp-3 |
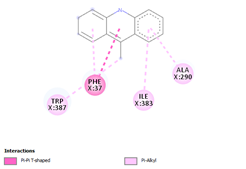 |
-8.1 |
1.138 |
|
Comp-4 |
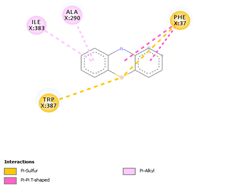 |
-7.2 |
5.206 |
|
Comp-5 |
 |
-7.6 |
2.648 |
|
Comp-6 |
 |
-10.8 |
0.012 |
|
Comp-7 |
 |
-10.0 |
0.046 |
|
Comp-8 |
 |
-10.0 |
0.046 |
|
Comp-9 |
 |
-3.9 |
1374.380 |
|
Comp-10 |
 |
-8.1 |
1.138 |
|
Comp-11 |
 |
-8.1 |
1.138 |
|
Comp-12 |
 |
-8.4 |
0.685 |
Comp-1 was docked with Ala268, Tyr239, Gln329, Leu150, and Phe154 at the binding pocket of the likely multidrug-efflux transporter Rv1258c. The binding affinity of Comp-1 was -6.7 kcal/mol. Comp-2 docked with Ala268, Tyr325, Phe154, Leu150, and Leu240. The binding affinities of Comp-2 and Comp-3 were -6.8 kcal/mol and -7.4 kcal/mol, respectively. The residues involved in the interaction were Leu240, Ala48, Met52, Phe154, Asp23, and Ile27. Comp-4 and Comp-5 showed binding affinities of -6.8 and -6.9 kcal/mol, respectively. The residues involved in these interactions were Ala268, Leu150, Phe154, Leu240, and Tyr325. Comp-6 showed the binding affinity of -8.7 kcal/mol. The residues involved in the interaction were Ala153, Ala20, Leu240, Leu150, and Asn151. The binding affinity of Comp-7 was -8.9 kcal/mol. The residues involved in the interaction were Ala20, Met52, Leu240, Leu150, Tyr239, and Phe154. The residues involved in the interaction for Comp-8 were Ala20, Met52, Leu240, Leu150, and Phe154. The binding affinity of Comp-8 was -8.4 kcal/mol. Comp-9 only interacted with a single residue of Tyr239 and the binding affinity was -3.7 kcal/mol. The binding affinity of Comp-10 was -7.4 kcal/mol. The residues involved in the interaction were Ala268, Phe154, Leu240, Tyr325, Tyr239, and Leu150. The binding affinities for Comp-11 and Comp-12 were -7.6 and 7.7 kcal/mol, respectively. The residues involved in these interactions were Ala268, Phe154, Tyr239, Leu150, and Leu240, respectively (Table 6).
Table 6. Binding Affinities of All Compounds with Probable Multidrug-efflux transporter Rv128c
|
Compound Name |
Interaction Diagram |
Binding Affinity (kcal/mol) |
Ki (µM) |
|---|---|---|---|
|
Comp-1 |
 |
-6.7 |
12.118 |
|
Comp-2 |
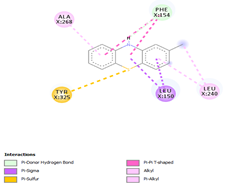 |
-6.8 |
10.234 |
|
Comp-3 |
 |
-7.4 |
3.713 |
|
Comp-4 |
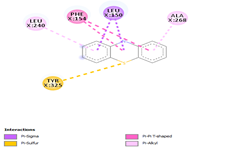 |
-6.8 |
10.234 |
|
Comp-5 |
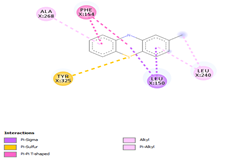 |
-6.9 |
8.643 |
|
Comp-6 |
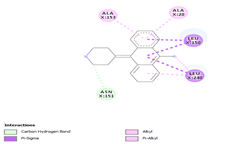 |
-8.7 |
0.413 |
|
Comp-7 |
 |
-8.9 |
0.294 |
|
Comp-8 |
 |
-8.4 |
0.685 |
|
Comp-9 |
 |
-3.7 |
1926.951 |
|
Comp-10 |
 |
-7.4 |
3.713 |
|
Comp-11 |
 |
-7.6 |
2.648 |
|
Comp-12 |
 |
-7.7 |
2.237 |
Comp-1 and Comp-3 were docked with Ala290, Ile383, Phe37, and Trp387 in the binding pocket of Rv1634, a potential multidrug-efflux transporter. Comp-1 and Comp-3 showed binding affinities of -7.2 kcal/mol and -8.2 kcal/mol, respectively. In the same way, the binding affinity of Comp-2 was -7.4 kcal/mol. The residues involved in the interaction were Leu318, Ala290, Ile383, Phe37, and Trp387. Comp-4 docked with Rv1634 at Thr156, Trp152, and Trp387, with a binding affinity of -7.2 kcal/mol. Comp-5 and Comp-6 were observed to dock with the binding affinity of -7.6 and -10.8 kcal/mol, respectively. The residues involved in these interactions were Trp152, Ala319, Trp322, Phe37, and Trp387. The binding affinity of Comp-7 was -10.0 kcal/mol. The residues involved in the interaction were Leu318, Ala319, Phe37, Thr156, Trp322, and Trp152. The residues involved in the interaction for Comp-8 were Ala290, Asp293, Leu318, Ala319, Phe37, Thr156, Trp322, Trp387, and Trp152. Comp-8 showed a binding affinity of -10.0 kcal/mol, while for Comp-9 it was -4.1 kcal/mol. Leu124, Tyr87, and Leu25 were the main residues involved in this interaction. Comp-10, Comp-11, and Comp-12 docked with Rv1634 at Trp152, Val29, Leu124 and Ala125, with a binding affinity of -8.2, -8.1, and 8.4 kcal/mol, respectively (Table 7).
Table 7. Binding Affinities of All Compounds with Probable Multidrug-efflux Transporter Rv1634.
|
Compound Name |
Interaction Diagram |
Binding Affinity (kcal/mol) |
Ki (µM) |
|---|---|---|---|
|
Comp-1 |
 |
-7.2 |
5.206 |
|
Comp-2 |
 |
-7.4 |
3.713 |
|
Comp-3 |
 |
-8.2 |
0.961 |
|
Comp-4 |
 |
-7.2 |
5.206 |
|
Comp-5 |
 |
-7.6 |
2.648 |
|
Comp-6 |
 |
-10.8 |
0.012 |
|
Comp-7 |
 |
-10.0 |
0.046 |
|
Comp-8 |
 |
-10.0 |
0.046 |
|
Comp-9 |
 |
-4.1 |
980.264 |
|
Comp-10 |
 |
-8.2 |
0.961 |
|
Comp-11 |
 |
-8.1 |
1.138 |
|
Comp-12 |
 |
-8.4 |
0.685 |
In silico analysis on absorption, distribution, metabolism, and excretion (ADME) was performed in terms of blood-brain barrier (BBB) permeability, plasma binding protein, skin permeability, and Caco-2 permeability (Supplementary S1).
3.6.1. Blood-Brain Barrier (BBB) Permeability. The blood-brain barrier (BBB) prevents the drug from entering the brain and eliminate toxicities with no side effects. It is determined in vivo as logBB values, which are logarithmic ratios of brain and plasma binding concentrations. Comp-1 had a BBB value of 1.9489, Comp-2 had a BBB value of 3.36813, Comp-3 had a BBB value of 4.70468, Comp-4 had a BBB value of 0.947887, Comp-5 had a BBB value of 1.33741, Comp-11 had a BBB value of 2.67734, and Comp-12 had a BBB value of 2.67404. Compounds with logBB > 0.3 are easily able to penetrate the blood-brain barrier, whereas compounds with logBB < -0.1 show low levels of penetration [30]. The compounds are all permeable to BBB.
3.6.2. Plasma Binding Protein. The high values of plasma binding protein predict that most of the drug is bound to the plasma protein, while the unbound part remains accessible for absorption. The quantity of binding to plasma proteins mainly affects the pharmacodynamics and pharmacokinetics of the drug. The unbound fraction of the drug to the plasma is calculated. In the case of HEPIs, the bound fraction for Comp-1, Comp-2, Comp-3, and Comp-8 was 100. For Comp-4 it was about 79.793057, for Comp-5 its value was 84.455782, for Comp-6 it was 88.972775, for Comp-7 it was 98.427317, for Comp-9 its value was 78.464943, for Comp-10 the value was 91.853135, for Comp-11 the value was 93.814698, and for Comp-12 it was 70.184825.
3.6.3. Skin Permeability. Skin permeability is vital for transdermal drug delivery. In vitro human skin permeability of 12 HEPIs was measured and the prediction was based on skin permeability constant logKp (cm/h). The logKp value for Comp-1 was -2.22865 cm/h, for Comp-2 it was -2.47242 cm/h, for Comp-3 it was -2.4654 cm/h, for Comp-4 it was -3.32973 cm/h, for Comp-5 it was -3.4087 cm/h, for Comp-6 it was -2.56731 cm/h, for Comp-7 it was -3.09064 cm/h, for Comp-8 it was -3.29524 cm/h, for Comp-9 it was -2.98568 cm/h, for Comp-10 it was -1.76497 cm/h, for Comp-11 it was -1.70922 cm/h, and for Comp-12 the value was -2.60175. A compound has high skin permeability if logKp < -2.5 and low skin permeability if logKp > -2.5.
3.6.4. Caco-2 Permeability. Human Colon Adenocarcinoma (Caco-2) permeability is regarded as an important factor in oral bioavailability. By using PreADMET, the Caco-2 permeability was measured and for Comp-1 the value was 26.7996 cm/sec. For Comp-2, the value was 16.3335 cm/sec, for Comp-3 it was 32.6339 cm/sec, for Comp-4 it was 26.9149 cm/sec, for Comp-5 it was 44.9944 cm/sec, for Comp-6 it was 28.0111 cm/sec, for Comp-7 it was 27.7414 cm/sec, for Comp-8 it was 47.145cm/sec, for Comp-9 it was 18.8133 cm/sec, for Comp-10 it was 28.628 cm/sec, for Comp-11 it was 42.8048 cm/sec, and for Comp-12 the value was 22.0097 cm/sec. A molecule has low Caco-2 permeability if the value is < 4. Whereas, if the value is between 4-70 then the molecule has medium permeability and if the value is > 70, then the molecule has high permeability. All the HEPIs of the current study were found to have medium permeability as all had their permeability values between 4-70.
3.6.5. Toxicity Analysis. Toxicity analysis was performed in terms of lethality and genotoxicity. The in-silico analysis was performed and the results were obtained. The lethal concentration (LC50) shows the concentration required to cause death in 50% of Fathead minnow. The value of LC50 is predicted as logLC50. When the value of LC50 is less than 0.5 mm, the compound is considered as high acute toxic [31]. The logLC50 value for Comp-1 was 0.00624363 mm. For Comp-2 it was 0.00194877, for Comp-3 the value was 0.00116833, for Comp-4 it was 0.0149294, for Comp-5 it was 0.00400254, for Comp-6 it was 0.00571874, for Comp-7 the value was 0.0271163, for Comp-8 the value was 0.012434, for Comp-9 the value was 0.0308489, for Comp-10 it was 0.0771713, and the values for Comp-11 and Comp-12 were 0.0430541 and 0.0799005, respectively. The hERG gene regulates the potassium channel and its inhibition leads to the development of acquiring QT syndrome, leading to a heart rhythm disorder. hERG inhibition for all the compounds was at medium risk, except Comp-11. However, the hERG inhibition for Comp-11 was at low risk. The mutagenic potential of the drug can be assessed by using the Ames test. The test was carried out by using PreADMET and the results showed that only Comp-6 and Comp-8 were non-mutagen, while all others were mutagen.
4. DISCUSSION
Tuberculosis (TB) continues to enhance morbidity in the world despite the use of curative chemotherapy. It is most likely to lead to human demise in developing countries [32, 33]. The WHO estimates that one third of the global population is infected with this contagious disease. Fortunately, anti-TB drugs are very effective and save around 35,000,000 lives in HIV negative patients. However, the struggle of control over TB is threatened due to the emergence of multidrug-resistant (MDR) strains of M. tuberculosis. Drug activation ezymes, such as efflux pumps (EPs) and their activity, may limit the effectiveness of the drug [18]. In order to make the antibiotics effective, different hybrid efflux pump inhibitors (HEPIs) were designed, since they sensitize the resistant strains of Mtb. In this study, a total of 12 HEPIs were evaluated in silico for their ability to interact with 5 Mtb EPs. The results demonstrated that HEPI-5, HEPI-7, and HEPI-11 consistently achieved strong binding affinities across multiple proteins, indicating their potential as broad-spectrum EP inhibitors. A large number of studies emphasize in vivo and in vitro analyses to develop drugs against TB. Mostly, it is preferred that an in silico analysis should be conducted before performing any experimental work. This type of analysis is cheap in terms of both time and cost.
The ADMET predictions further refined the assessment of lead compounds. Importantly, HEPI-5, HEPI-7, and HEPI-11 combined favorable docking with good oral absorption, non-mutagenicity, and acceptable toxicity profiles. However, potential hepatotoxicity flagged in HEPI-3 and HEPI-8 warrants caution, as hepatotoxicity is a major limitation in TB chemotherapy [34]. LC₅₀ values for most compounds fell within low-to-moderate toxicity ranges when compared with published zebrafish embryo toxicity thresholds [35], supporting their potential safety but underscoring the need for in vivo validation.
The blood-brain barrier (BBB) predictions also have important clinical implications. For pulmonary TB, CNS penetration may not be essential and low BBB permeability could reduce the neurotoxicity risk. However, for TB meningitis—a severe form of extrapulmonary TB—CNS penetration is critical [36]. Some predicted values appeared unusually high (>4), which have been clarified as limitations of the computational model. Future optimization should focus on balancing systemic exposure with controlled CNS penetration depending on the intended clinical usage.
This study identified HEPI-5, HEPI-7, and HEPI-11 as promising scaffolds that outperform weaker candidates (e.g., HEPI-2 and HEPI-4), both in docking strength and ADMET properties. Compared to existing inhibitors [37], these compounds showed the advantage of a hybrid structural design, offering better binding consistency across multiple EPs. Their broader significance lies in providing a foundation for developing novel EPIs that could be co-administered with standard anti-TB drugs to restore their efficacy against resistant strains.
4.1 Limitations of the StudyThis study was limited to in silico predictions. Moreover, the reliability of docking outcomes depends on the accuracy of the homology models and the scoring functions used. Although ADMET tools provided valuable insights, these tools cannot fully model the complexity of drug metabolism, toxicity, and blood-brain barrier permeability in vivo. Furthermore, the absence of experimental validation means that biological activity, safety, and pharmacokinetics of the proposed compounds remain unconfirmed.
4.2. ConclusionThis study identified HEPI-5, HEPI-7, and HEPI-11 as the most promising HEPIs against M. tuberculosis. These compounds consistently showed strong binding affinities across multiple EPs, favorable pharmacokinetic properties, and acceptable predicted toxicity profiles. Compared with existing inhibitors, these candidates showed the advantage of a hybrid structural design, providing enhanced binding and more favorable ADMET characteristics. Importantly, the assessment of BBB permeability and toxicity profiles placed the findings in the clinical context of pulmonary TB and TB meningitis, where safety and CNS penetration requirements differ. Thus, it is concluded that these HEPIs can cause effective TB inhibition in human beings. However, some of the pharmacological properties are non-suitable for humans which needs to be validated in vitro and in vivo before their administration to human beings.
CONFLICT OF INTEREST
The authors of the manuscript have no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
DATA AVAILABILITY STATEMENT
Data associated with this study will be provided by corresponding author on reasonable request.
FUNDING DETAILS
The authors did not receive any funding for this article.
REFERENCES
- Olmo-Fontánez AM, Turner J. Tuberculosis in an aging world. Pathogens. 2022;11(10):e1101. https://doi.org/10.3390/pathogens11101101
- Alland D, Kalkut GE, Moss AR, et al. Transmission of tuberculosis in New York City--an analysis by DNA fingerprinting and conventional epidemiologic methods. N Engl J Med. 1994;330:1710-1716. https://doi.org/10.1056/NEJM199406163302403
- World Health Organization. Global tuberculosis report 2018. WHO Web site. https://www.who.int/publications/i/item/9789241565646
- Song Y, Li T, Xia H, et al. Analysis of the epidemiological characteristics of national reported pulmonary tuberculosis incidence, 1997—2023. Chin J Antituberc. 2024;46(10):1198-1208. https://doi.org/10.19982/j.issn.1000-6621.20240382
- Falzon D, Schünemann HJ, Harausz E, et al. World Health Organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur Respir J. 2017;49(3):e1602308. https://doi.org/10.1183/13993003.02308-2016
- Giurazza R, Mazza MC, Andini R, Sansone P, Pace MC, Durante-Mangoni E. Emerging treatment options for multi-drug-resistant bacterial infections. Life. 2021;11(6):e519. https://doi.org/10.3390/life11060519
- Alam A, Locher KP. Structure and mechanism of human ABC transporters. Annu Rev Biophys. 2023;52(1):275-300. https://doi.org/10.1146/annurev-biophys-111622-091232
- Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuberc Lung Dis. 1998;79(1):3-29. https://doi.org/10.1054/tuld.1998.0002
- Belay WY, Getachew M, Tegegne BA, et al. Mechanism of antibacterial resistance, strategies and next-generation antimicrobials to contain antimicrobial resistance: a review. Front Pharmacol. 2024;15:e1444781. https://doi.org/10.3389/fphar.2024.1444781
- Huang L, Wu C, Gao H, et al. Bacterial multidrug efflux pumps at the frontline of antimicrobial resistance: an overview. Antibiotics. 2022;11(4):e520. https://doi.org/10.3390/antibiotics11040520
- Henderson PJ, Maher C, Elbourne LD, Eijkelkamp BA, Paulsen IT, Hassan KA. Physiological functions of bacterial “multidrug” efflux pumps. Chem Rev. 2021;121(9):5417-5478. https://doi.org/10.1021/acs.chemrev.0c01226
- Lake MA, Adams KN, Nie F, et al. The human proton pump inhibitors inhibit Mycobacterium tuberculosis rifampicin efflux and macrophage-induced rifampicin tolerance. Proc Natl Acad Sci. 2023;120(7):e2215512120. https://doi.org/10.1073/pnas.2215512120
- Van Bambeke F, Glupczynski Y, Plesiat P, Pechere J, Tulkens PM. Antibiotic efflux pumps in prokaryotic cells: occurrence, impact on resistance and strategies for the future of antimicrobial therapy. J Antimicrob Chemother. 2003;51(5):1055-1065. https://doi.org/10.1093/jac/dkg224
- AlMatar M, Albarri O, Makky EA, Köksal F. Efflux pump inhibitors: new updates. Pharmacol Rep. 2021;73:1-16. https://doi.org/10.1007/s43440-020-00160-9
- Sreekantan AP, Rajan PP, Mini M, Kumar P. Multidrug efflux pumps in Bacteria and efflux pump inhibitors. Adv Microbiol. 2022;61(3):105-114. https://doi.org/10.2478/am-2022-009
- Laws M, Jin P, Rahman KM. Efflux pumps in Mycobacterium tuberculosis and their inhibition to tackle antimicrobial resistance. Trends Microbiol. 2022;30(1):57-68. https://doi.org/10.1016/j.tim.2021.05.001
- Pieroni M, Machado D, Azzali E, Costa SS, Couto I, Costantino G, Viveiros M. Rational design and synthesis of thioridazine analogues as enhancers of the antituberculosis therapy. J Med Chem. 2015;58(15):5842-5853. https://doi.org/10.1021/acs.jmedchem.5b00428
- Breijyeh Z, Karaman R. Design and synthesis of novel antimicrobial agents. Antibiotics. 2023;12(3):e628. https://doi.org/10.3390/antibiotics12030628
- Ch'ng JH, Mok S, Bozdech Z, et al. A whole cell pathway screen reveals seven novel chemosensitizers to combat chloroquine resistant malaria. Sci Rep. 2013;3:e1734. https://doi.org/10.1038/srep01734
- Germann UA, Harding MW. Chemosensitizers to overcome and prevent multidrug resistance? Chin Chem Lett. 2025;36(1):e109724. https://doi.org/10.1016/j.cclet.2024.109724
- Yu S, Zhang S, Zhang A, Han J, Sun B. Design, Synthesis, and activity evaluation of novel Bifenamide dual-target antibacterial inhibitors and carrier based on infectious microenvironment. J Med Chem. 2025;68(4):4743-4762. https://doi.org/10.1021/acs.jmedchem.4c02913
- Asha IJ, Gupta SD, Hossain MM, et al. 2024. In silico characterization of a hypothetical protein (PBJ89160. 1) from Neisseria meningitidis exhibits a new insight on nutritional virulence and molecular docking to uncover a therapeutic target. Evol Bioinform. 2024;20:1-6. https://doi.org/10.1177/11769343241298307
- Altunkulah E, Ensari Y. Protein structure prediction: an in-Depth comparison of approaches and tools. Eskişehir Tech Univ J Sci Technol. 2024;13(1):31–51. https://doi.org/10.18036/estubtdc.1378676
- Kesheri M, Kanchan S, Häder DP, Sinha RP. Proteomics and bioinformatics approaches for exploring resilience strategies in Cyanobacteria. In: Kesheri M, Kanchan S, Häder DP, Sinha RP, eds. Multi-Omics in Biomedical Sciences and Environmental Sustainability. Springer Nature; 2025:407-427. https://doi.org/10.1007/978-981-96-7067-3_17
- Alhusayni AA. The role of AutoDock and related techniques in bioinformatics for developing immune proteins and therapeutic treatments. Infin J Med Innovat. 2025;1(3):62-66.
- Tang S, Ding J, Zhu X, et al. Vina-GPU 2.1: towards further optimizing docking speed and precision of AutoDock Vina and its derivatives. IEEE/ACM Transac Comput Biol Bioinfo. 2024;21(6):2382-2393. https://doi.org/10.1109/TCBB.2024.3467127
- Nelapati AK, Meena SK. An approach to increase the efficiency of uricase by computational mutagenesis. Phys Chem Res. 2023;11(3):481-491. https://doi.org/10.22036/pcr.2022.345329.2115
- Taranto AG, Junior MC. Tools and techniques in structural bioinformatics. In Singh TR, Saini H, Junior MC, eds. Bioinformatics and Computational Biology. Chapman and Hall/CRC; 2023:173-185. https://doi.org/10.1201/9781003331247
- Lee SK, Park SH, Lee IH, No KT. PreAD-MET Ver. v2. 0. BMDRC: 2007.
- Al-Jarf R, de Sá AG, Pires DE, Ascher DB. pdCSM-cancer: using graph-based signatures to identify small molecules with anticancer properties. J Chem Inf Model. 2021;61(7):3314-3322. https://doi.org/10.1021/acs.jcim.1c00168
- Mvondo JG, Matondo A, Mawete DT, Bambi SM, Mbala BM, Lohohola PO. In silico ADME/T properties of quinine derivatives using SwissADME and pkCSM webservers. Int J Trop Dis Health. 2021;42(11):1-2. https://doi.org/10.9734/IJTDH/2021/v42i1130492
- Mohammadnabi N, Shamseddin J, Emadi M, et al. Mycobacterium tuberculosis: the mechanism of pathogenicity, immune responses, and diagnostic challenges. J Clin Lab Anal. 2024;38(23):e25122. https://doi.org/10.1002/jcla.25122
- Abdel-Halim MS, El-Ganiny AM, Mansour B, Yahya G, Latif HK, Askoura M. Phenotypic, molecular, and in silico characterization of coumarin as carbapenemase inhibitor to fight carbapenem-resistant Klebsiella pneumoniae. BMC Microbiol. 2024;24:e67. https://doi.org/10.1186/s12866-024-03214-7
- Zade D. Hepatotoxicity associated with anti-tuberculosis medications: analyzing mechanisms, risk factors, and strategies for prevention and management. J Drug Deliv Biotherap. 2024;1(3):1-12. https://doi.org/10.61920/jddb.v1i03.155
- Bertoni Í, Sales BC, Viriato C, Peixoto PV, Pereira LC. Embryotoxicity induced by triclopyr in zebrafish (danio rerio) early life stage. Toxics. 2024;12(4):e255. https://doi.org/10.3390/toxics12040255
- Madadi AK, Sohn MJ. Comprehensive therapeutic approaches to tuberculous meningitis: Pharmacokinetics, combined dosing, and advanced intrathecal therapies. Pharmaceutics. 2024;16(4):e540. https://doi.org/10.3390/pharmaceutics16040540
- Rodrigues L, Cravo P, Viveiros M. Efflux pump inhibitors as a promising adjunct therapy against drug resistant tuberculosis: a new strategy to revisit mycobacterial targets and repurpose old drugs. Expert Rev Anti Infect Ther. 2020;18(8):741-757. https://doi.org/10.1080/14787210.2020.1760845