Optimized Spectral Clustering Methods For Potentially Divergent Biological Sequences
Johny Matar1, 2*, Hicham EL Khoury2, Jean-Claude Charr1, Christophe Guyeux1, and Stéphane Chrétien3
1Université de Bourgogne Franche-Comté, Besançon, France
2LaRRIS, Faculty of Sciences, Lebanese University, Fanar, Lebanon
3Laboratoire ERIC, Université Lyon 2, Bron, France
Abstract
Various recent researches in bioinformatics demonstrated that clustering is a very efficient technique for sequence analysis. Spectral clustering is particularly efficient for highly divergent sequences and GMMs (Gaussian Mixture Models) are often able to cluster overlapping groups if given an adequately designed embedding. The current study used spectral embedding and Mixture Models for clustering potentially divergent biological sequences. The research approach resulted in a pipeline consisting of the following four steps. The first step consists of aligning the biological sequences. The pairwise affinity of the sequences is computed in the second step. Then the Laplacian Eigenmap embedding of the data is performed in the third step. Finally, the last step consists of a GMM-based clustering. Improving the quality of the generated clustering and the performance of this approach is directly related to the enhancement of each one of these four steps. The main contribution is proposing four GMM-based algorithms for automatically selecting the optimal number of clusters and optimizing the clustering quality. A clustering quality assessment method, based on phylogenetic trees, is also proposed. Moreover, a performance study and analysis have been conducted while testing different clustering methods and GMM implementations. Experimental results demonstrated the superiority of using the BIC (Bayesian Information Criterion) for selecting the optimal GMM configuration. Significant processing speed improvements were also recorded for the implementation of the proposed algorithms.
1. Introduction
In the last two decades, the cost of sequencing a genome has decreased at a dramatic rate from 100 million dollars at the end of the 20th century to less than $1000 in the present times. This led to an explosion in the number of sequenced genomes and proteins. The large number of newly discovered biological sequences allowed the researchers in bioinformatics to study the relationships between the different sequenced species and to reconstruct their phylogenetic tree and ancestors. Many tools were developed to analyze the sequenced data. In particular, clustering packages were implemented to compare a set of sequences and regroup them into clusters according to their similarity. However, it is worth emphasizing that mutations can lead to similarities lower than 85% between sequences belonging to the same cluster [1].
The clustering of biological sequences is currently playing a paramount role in linking the huge number of newly discovered sequences to their variants and ancestors. However, current methods can only partially tackle this problem due to its scale and complexity. Several research works [2–4] have concluded that spectral clustering may represent an efficient tool for biological sequence clustering and, to our knowledge, only one of them has been publicly released [4]. In [4], the relevance of using GMMs (Gaussian Mixture Models) for unsupervised clustering of biological sequences was demonstrated through various numerical validation experiments. Contrarily, to most of the widely used clustering tools, GMM-based approaches require no user intervention and are well adapted to clustering divergent sequences as well. The targeted sequences could be mutations from a same gene (or genome), or even cross-species divergent but homologous sequences or fragments.
The difficulty in studying newly discovered biological sequences lies primarily in their unknown degree of divergence when compared to each other or to other known sequences. Therefore, neither the accurate selection of the similarity threshold nor the selection of the clusters' centroids is trivial for a traditional clustering tool. In such cases, traditional tools, requiring a user-defined similarity threshold, cannot be considered reliable. On the other hand, GMM-based alternatives which do not require any a priori knowledge of an arbitrary similarity threshold, seem to be well adapted to efficiently tackle such problems. GMM showed good classification performances in several applications where clusters overlap, such as biological sequence clustering [4], age and gender recognition [5], real-time segmentation of HD video [6], and others. GMMs and other similarly finite mixture models [7] are usually calibrated using an Expectation Maximum (EM) algorithm [8–10] or one of its accelerations [11–13]. However, the use of EM-type algorithms requires expertise due to the well-known drawbacks [14, 15] and computational issues for large and high dimensional data [16]. Therefore, users should rely on packages that carefully address these subtle technical issues.
The tool presented in [4] implements the following operations for clustering a set of biological sequences. These include, (i) sequences' alignment, (ii) pairwise affinity computation of the sequences, (iii) Laplacian eigenmap embedding of the data, and (iv) GMM-based clustering. The quality of the generated clustering and the performance of this approach are often greatly impacted by the tool or the algorithm used at each stage. These tools or algorithms affect the alignment quality, the pairwise similarity computation between sequences, and the GMM performance. The current study investigates how the use of different techniques and their implementations at the clustering stage contribute to accelerating the clustering or improving its quality. Our contributions include suggesting a significantly faster substitute to the GMM that was used in [4], while proposing new GMM-based algorithms for enhancing the quality of the clustering. The experimented features, methods, and algorithms were integrated into a clustering package published on a public online repository[1].
The remainder of this article is organized as follows. In Section 2, the clustering of biological sequences is introduced and different clustering techniques are detailed. The sequences' alignment, the affinity computation methods, and some existing GMM implementations are also presented in this section. In Section 3, four approaches to automatically choose the most relevant clustering based on given criteria are presented. The experimental protocol is detailed in Section 4. The results of the experiments are presented and discussed in Section 5. Finally, Section 6 recapitulates the findings and Sub Section 6.1 presents some future prospects for the current project.
2. STATE-OF-THE-ART
2.1. Clustering Biological Sequences
Several researches were conducted to efficiently cluster biological sequences. However, most of the proposed approaches are highly sensitive to user-defined parameters, that is the similarity or identity threshold. Moreover, they are designed to quickly cluster highly similar sequences. Indeed, the lowest possible similarity threshold is usually larger than 75% (for example, in tools, such as CD-HIT-EST) and most of the experiments conducted in the studies introducing these tools, only consider similarities larger than 85%. These tools are not able to accurately detect communities among potentially divergent sequences. To sum up, existing clustering packages can be broadly divided into two categories based on their objectives, which are as follows:
- Packages and tools suitable for fast clustering of highly similar sequences but requiring a user-defined threshold.
- Intervention-free tools that can even cluster potentially divergent sequences.
The most popular algorithms and tools from both categories are presented in the next subsections.
2.1.1. Fast Clustering of Highly Similar Sequences>. High-speed clustering of highly similar sequences mostly relies on greedy, hierarchical, Dirichlet Process means (DP-means) [17], or mean shift [18] algorithms. It requires some user-defined parameters, such as a similarity or identity threshold and, optionally, the centroids of the clusters. The sequences are then grouped into clusters based on the provided parameters. Following this scheme, several tools are publicly available, such as CD-HIT [19], UCLUST [20], DNACLUST [21], HPC-CLUST [22], and DACE [23]. Most of these tools group the sequences around the clusters representatives or centroids based on a user-provided similarity threshold, but they differ in the way they choose these representatives.
CD-HIT and DNACLUST order the sequences according to their length. Each sequence is either added to a previously created cluster, if its similarity with a previously chosen centroid does not exceed the user-provided threshold or it is considered as a new centroid for a new cluster. In contrast, UCLUST performs the classification without prior sorting of the input sequences, thus, the input order might impact the resulting clustering.
In order to achieve better clustering speed, various approaches were adopted by the aforementioned tools. CD-HIT avoids the costly pairwise sequences' alignment by using word counting for computing similarities. HPC-CLUST takes an already aligned set of sequences as input and it uses a distributed hierarchical algorithm that clusters subsets of the sequences and finally merges the closest clusters. DACE uses parallel computation to rapidly cluster large datasets. After an iterative partitioning of the input sequences into non-intersecting subsets, DACE uses the DP-means algorithm to cluster the sequences in parallel.
The type of supported input sequences represents another distinction between the clustering tools. The CD-HIT package offers CD-HIT-EST for nucleotide sequences clustering and CD-HIT-PROTEIN for protein sequences clustering, while UCLUST and DACE can cluster both types of sequences. Conversely, DNACLUST and HPC-CLUST are not designed to handle protein sequences.
2.1.2. Intervention-free Clustering of Potentially Divergent Sequences. The mutations in biological sequences occur in variable and unpredictable degrees which eventually turn the choice of the identity or similarity threshold into a challenging dilemma when clustering a set of sequences without a priori knowledge. Only a few recent studies tackled this problem and were successful in clustering potentially divergent sequences [4, 24]. Their solutions relied on mixture models and performed the clustering based on a probability distribution [25]. Contrary to the tools targeting highly similar sequences mentioned above, these packages do not need any user intervention, especially for the choice of the identity or similarity threshold.
The authors in [24] proposed an original Python-based clustering package that uses an unsupervised learning approach, namely the Gaussian Mixture Model clustering applied after a Laplacian Eigenmap dimensionality reduction. It is noted that the Gaussian Mixture Model [26] is a probabilistic model for detecting sub-communities within a certain community. The objective of this package is accurate clustering even for divergent sequences. The number of clusters is determined using statistical criteria, such as the Bayesian information criterion (BIC) [27]. The use of this statistical criterion leads to an autonomous process that does not rely on neither user-chosen clusters' centroids nor identity thresholds. This clustering package consists of four main stages.
- Sequence Alignment: This stage relies on the third-party module MUSCLE [28] to align the sequences.
- Similarity Matrix Calculation: An NxN square matrix, where N is the number of input sequences and each (i,j) element is the pairwise similarity index between sequences i and j. Similarity indices are derived from the pairwise distances between sequences that are computed with the EDNAFULL scoring matrix.
- Dimensionality Reduction: The Laplacian Eigenmap of a transformed version of the similarity matrix, known as the affinity matrix, is computed, leading to a size reduction of the matrix.
- Sequence Clustering: In this last stage, the Gaussian Mixture Model is applied to the results obtained in Step 3 to cluster the sequences.
This model exhibited competitive results, especially in the case of highly divergent sequences. However, its speed significantly degraded when applied to large datasets.
Since the similarity matrix calculation stage represents an intensive computation step of the order of O((N2-N)/2), the authors in [4] proposed an optimized hybrid C++/Python package where the second stage is implemented in C++ and computed in parallel to reduce its execution time. Based on the experimental results published in [4], the hybrid package delivers up to 126X speed-up when compared to the original package. Additionally, its capabilities were extended to cluster protein sequences by introducing two additional scoring matrices, namely BLOSUM62 and PAM250 [29].
Despite the advantageous intervention-free property of the latter algorithm and its performance improvement, it is still not expected to scale well for large datasets. This is due to the alignment required in its first stage. Conversely, further accuracy and speed improvements remain possible by enhancing each one of its stages. In the next three subsections, the possible improvements for each stage are discussed.
2.2. Sequences' Alignment and Similarity Computation
One of the fundamental techniques for visualizing the dissimilarities and computing the distance between a pair of sequences is their alignment. This technique discloses the mutations, insertions, and deletions phenomena that differentiate the sequences. Therefore, many efficient algorithms were proposed for aligning the sequences and computing the pairwise distances, such as Needleman-Wunsch, Sankoff, and Sellers [30]. MUSCLE [28], MAFFT [31], DECIPHER [32], and CLUSTALX [33] are a few examples of alignment tools. The alignment speed and accuracy represent two major differentiating aspects between these tools that might influence the clustering quality. Therefore, it is crucial to investigate the effects of the alignment on the spectral clustering technique in order to enhance the quality of the produced clustering.
2.3. Affinity Matrix Computation
Following the alignment, the pairwise distance is computed using a string metric, such as the Needleman-Wunsch distance. Then, the similarity is inferred from the pairwise distance. For instance, in [21], the similarity is equal to:
1-" distance " /" length of the shorter sequence "
The distance choice and the similarity definition vary from package to package, which might produce different clusterings, even when considering the same similarity threshold.
In [24] and [4], the affinity matrix was computed as a Random Walk Normalized Laplacian and it proved to be relevant for the clustering of biological sequences. However, other interesting matrices have been proposed for spectral clustering [34–37], such as the Non-normalized Laplacian, Modularity [35], and the Bethe Hessian (Deformed Laplacian) [38]. These matrices are defined as follows:
- Non-normalized Laplacian:
L=D-A
where A is the adjacency matrix between the sequences and is its diagonal matrix of degrees.
- Random Walk Normalized Laplacian:
L^rw=D^(-1) L
where is the degrees matrix of the adjacency matrix and is the Non-normalized Laplacian matrix. The Laplacian matrix is symmetric and positive semidefinite.
- Modularity:
M=1/K (A-1/K kk^T )
where is the adjacency matrix, is the degrees vector of , and is the total degree of . High values for this quality function reveal the possible existence of strong communities.
- Bethe Hessian:
H_r=(r^2-1)I+D-rA
where is the identity matrix, is the degrees matrix of the adjacency matrix , and the constant is the square root of the average degree of the graph, as suggested in [36].
2.4. The GMM Implementations
The last stage of this spectral clustering tool uses the GMM. Various implementations of this mixture model are publicly available, such as the GaussianMixture() [39] and spectral_embedding() [40] functions from Python's scikit-learn library [41]. Moreover, there are also free and standalone[1] C++ implementations of the GMM, such as the paperrune [42] and our implementation[2][43]. The GMMs implemented with a lower-level programming language (C++ vs Python) are expected to compute faster and enhance the speed and the scalability of this heavy-computational approach.
Most of the implementations of GMM take an features matrix[3] as input, where is the number of features and is the number of samples. Conversely, spectral_embedding() [40] merges the dimensionality reduction and the sequence clustering phases and takes an pairwise similarity matrix as input, where is the number of samples. The (normalized or not) Laplacian matrix computation is embedded in the spectral_embedding() function. The number of resulting clusters can be specified in the parameter described as dimension of the projection subspace. By default, this parameter is set to 8.
The above-mentioned libraries do not exactly apply the same algorithms and, therefore, they do not give identical results. Moreover, these implementations do not offer the same features. For instance, some of them include the computation of some information criteria that reflect the quality of GMM [44], such as the Log-Likelihood implemented in [42], or the Bayesian Information Criterion (BIC) implemented in [43] and [39]. The BIC is defined as follows:
BIC=ln(n)k-2ln(L)
where is the data size, is the number of features for the model, and is its likelihood. The spectral_embedding() function does not provide any method to compute statistical indices of quality. These statistical indexes can be exploited to improve the produced clustering.
These libraries could also take a seed as input that affects the initial random distribution and might possibly affect the resulting clustering. If not provided, this seed is randomly generated. Since, the C++ pseudorandom number generator, which is used to generate the random seeds in the GMM implementations, is not cross-platform consistent[6] , therefore a custom pseudorandom generator was introduced in [43] in order to preserve the consistency of the results. It is based on Microsoft's Rand formula:
(a*seed+c)%m where a=214013, c=2531011, and m=2^31
If no seed is provided by the user, the seed is equal by default to 0. In the next section, the main contributions of this work are presented.
3. APPROACHES AND METHODS
3.1. Four Approaches to Fine-tuning the GMM
Given the promising advantages of the spectral clustering in the aforementioned tools [4, 24], the study's approach is to exploit the parameters of the state-of-the-art GMM implementations, to fine-tune the produced clusterings and improve their quality. Four approaches to automatically select the most relevant clustering based on given criteria are presented in this section. The first algorithm consists of maximizing the GMM likelihood. This is achieved by performing several iterations as illustrated in Figure 1.
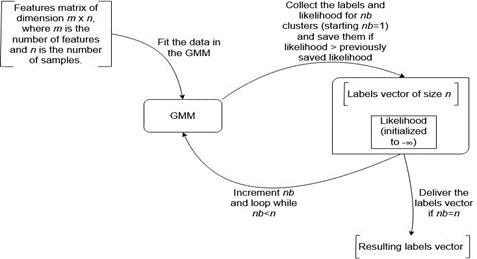
Figure 1. Choosing the Best Clustering Based on Maximum Likelihood
The given number of clusters is modified at each iteration and it ranges between 1 and the number of sequences. The second approach is similar to the previous one. It simply substitutes the maximum likelihood with the lowest BIC. It is worth noting that additional implementations using the AIC (Akaike Information Criterion) and ICL (Integrated Complete Likelihood) were omitted because they resulted in the same output when compared to the implementation using BIC.
The third approach consists of executing the previous algorithm for a user-defined number of times, with a different random seed at each iteration. It is important to recall that the random seed impacts the initial random distribution of the centroids, leading to a potentially different clustering for each seed. The clustering that scores the maximum number of occurrences is selected. The counting procedure for the occurrences of each clustering distinguishes between the same clustering with different labeling and different clustering. Figure 2 illustrates this method.
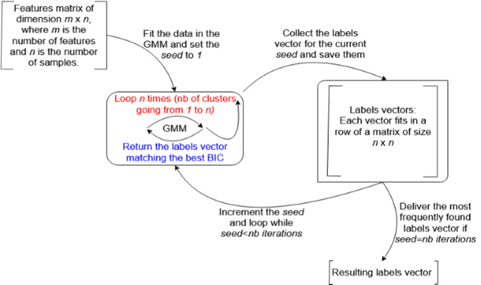
Figure 2. Choosing the Best Clustering Based on the Occurrence Frequency
The computation time of this approach, compared to the previous one, is proportional to the chosen number of iterations. Moreover, this algorithm requires a larger amount of memory, since it saves the labels vector for the resulting clustering at each iteration. Therefore, it requires a substantial amount of memory if the input dataset and the chosen number of iterations are both large.
The fourth algorithm shares some aspects of similarity with the third one. It successively clusters the sequences using different seeds, but just keeps in memory the designated best clustering (for example, the one that scores the best BIC). Moreover, in order to reduce the execution time of this algorithm, an additional parameter, called noImp, is defined to count the number of iterations made without detecting any improvement and stop the iterative process before reaching the chosen number of iterations (nb iterations). For example, the iterative process is terminated if no BIC improvement is detected after a user-defined maxNoImp number of consecutive iterations (noImp=maxNoImp).
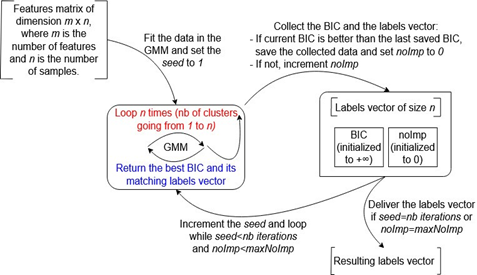
Figure 3. Choosing the Best Clustering Based on the Best Reached BIC
Figure 3 illustrates this algorithm that requires less computation time than the previous one in the case where the execution stopping condition is fulfilled prior to reaching the chosen number of iterations. The detailed inputs and parameters, for the implementations that were used in these four algorithms, can be found in Tables 4 and 5 in Appendix. To evaluate the four methods on real datasets, where a clustering ground truth is unknown, an additional method for selecting a reference clustering is proposed in the next subsection.
3.2. Generating a Reference Clustering
When the properties of a certain set of sequences are unknown, establishing the evolutionary relationship between these sequences is a challenging step. This relationship can be represented by a phylogenetic tree that helps in individually assessing each clustering of the dataset. Since it is possible in each clustering to identify valid subclusters, it is not fair to assess all the clusterings by using a single unified reference per dataset. Therefore, a custom algorithm is defined for assigning a reference for each produced clustering. Primarily, this algorithm aims to define a reference clustering, that is based on an existing clustering and in which a certain acceptable cluster is supposed to fully cover a sub-branch in the phylogenetic tree. The algorithm receives as inputs, a certain clustering along with the phylogenetic tree of the clustered sequences. It produces a reference clustering based on the input. This algorithm consists of the following steps.
- From the given clustering, the elements of the phylogenetic tree are assigned labels as illustrated in Figure 4. The labels indicate to which cluster each sequence belongs in the given clustering. For example, in Figure 4, the clustering produced four clusters where clusters 1 to 4 are represented by the labels *, #, -, and + respectively.
- The depth of the phylogenetic tree (TD) is computed and a counter is initialized to . At each iteration, it is decremented by 1 till it reaches 0.
- On each iteration, for each inner node that has a depth equal to the counter, the following cases are possible.
a) If all the first-level descendants of the node are leaves, a cluster consisting of such leaves is formed. The newly formed cluster is labeled according to the dominant label, the label that occurs the most among the cluster elements. If no dominant label was found, that is two labels have the same high number of occurrences, the undefined label is attributed to the cluster.
b) If the first-level descendants of the node include a leaf and at least one already formed cluster, the leaf is added to the cluster that is the closest to it. The cluster is relabeled according to the dominant label between its elements.
c) If the first-level descendants of the node form at least two clusters, the sub-tree under this node is processed as follows:
I. any couple of adjacent clusters having the same label are merged,
II. if a cluster is labeled as "undefined", it is merged with an adjacent cluster and the resulting cluster is relabeled according to the new dominant label between its elements,
III. if two adjacent clusters have different labels, they are not modified, and
IV. each small cluster left (having less than 4 elements), if any, is merged with its surrounding larger clusters, in case these surrounding clusters have the same label.
4. After the final iteration, if there are still clusters with undefined labels, they are assigned new labels. If two or more clusters have the same label, they are also assigned new labels.
Figures 4 and 5 illustrate how a reference clustering is generated according to the algorithm described above. In the first sub-figure of Figure 4, the elements of the phylogenetic tree are assigned labels or +), that indicate to which cluster each sequence belongs in the given clustering. The depth of each node in the tree is also displayed. In this example, the depth of the tree is equal to 6. After this initialization step, the iterative process starts with the inner nodes at depth . The second subfigure of Figure 4 illustrates the first iteration of the algorithm. In this example, there is only one inner node with a depth . It contains two leaves/sequences (Elt 11 and Elt 12). Both sequences belong to the third cluster. Therefore, a cluster containing both sequences is formed and labeled as "Cluster 3" in the reference clustering. This new cluster is represented by a red rectangle in Figure 4 whereas Figure 5 illustrates the remaining iterations.
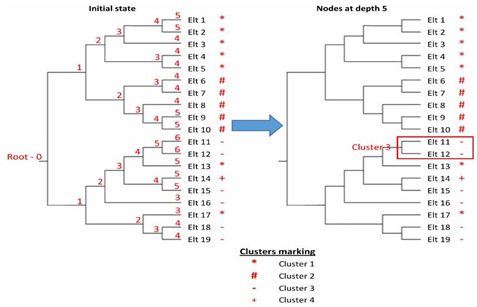
Figure 4. Initial State and First Iteration
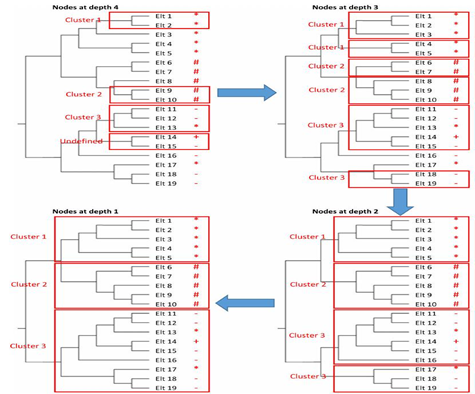
Figure 5. Clusters Identification and Final State
At the second iteration with inner nodes of depth , three new clusters are created. The first one consists of Elt 1 and Elt 2 and is labeled as "Cluster 1" because both of its sequences belong to the first cluster. The second cluster is created in the same way as the previous one. The third new cluster consists of Elt 14 and Elt 15 which belong to different clusters and thus, there is no dominant label in this cluster. For this reason, the third identified cluster is labeled as "Undefined". It can also be noticed that Elt 3 was added to "Cluster 3" and the label of this cluster is preserved since the dominant label among its elements did not change. Figure 5 displays the next three iterations and then the iterative process stops at the root node depth . In this scenario, the resulting reference clustering consists of three clusters. The first two are homogeneous but the third one contains sequences belonging to three different clusters in the given clustering. However, six of its nine sequences belong to the same cluster and thus, their dominant label is assigned to this cluster.
4. EXPERIMENTAL PROTOCOL
4.1. Datasets
Three real biological sequence datasets have been considered to evaluate the proposed approaches in the spectral clustering pipeline. They are as follows:
- A first set of 78 complete genome sequences, belonging to HIV-1 type B virus samples identified in Cyprus and downloaded from the Los Alamos National Laboratory's website[7].
- A second set of 100 genomic sequences, belonging to the NADH dehydrogenase 3 (ND3) mitochondrial gene, from a collection of Platyhelminthes and Nematoda species.
- A third set of 24 different nucleoprotein (NP) sequences, belonging to the strain A/H1N1 of the Influenza virus and downloaded from NCBI's Influenza Virus database[8].
Table 1 shows a brief description of the first three datasets which contain each a single type of sequences. The statistics on the sequences were retrieved from the output of MUSCLE [28]. The pairwise similarity, between the sequences of each dataset, was computed using MatGAT [45] which calculates the similarity after using the Myers and Miller global alignment algorithm [46].
Table 1. Statistical Description of the Real Datasets
|
Datasets |
Seqs Count |
Max Length |
Avg Length |
Min Similarity |
Max Similarity % |
Avg Similarity % |
|
HIV |
78 |
8272 |
8167 |
86 |
99.4 |
89.6 |
|
NADH |
100 |
369 |
341 |
46.2 |
99.7 |
62.8 |
|
Influenza |
24 |
498 |
498 |
97.4 |
99.8 |
98.8 |
Since a clustering ground truth is not available for these three datasets, a phylogenetic tree, showing the evolutionary relationship among the sequences of each set, is used for producing individual reference clusterings later, based on the proposed method in Section 3. Indeed, there are many tools that, given an aligned set of sequences, can build the phylogenetic tree of these sequences. In this work, the tree for each set of data was built according to the following procedure.
- MUSCLE [28] computed the sequences' alignment.
- PhyML 3.0 [47] generated the phylogenetic tree. The automatic model selection, based on the likelihood criteria, was selected. This selection, provided by SMS [48], was set to use the BIC (Bayesian Information Criterion).
- The resulting phylogenetic tree was visualized using PRESTO (Phylogenetic tReE viSualisaTiOn[9]).
All these assembled datasets are publically hosted on an online repository[10].
4.2. Experiments
The current research's applied set of experiments aims to compare the GMM implementations presented in Section 2 and the GMM-based algorithms proposed in Section 3. The three first datasets were used for this set of experiments. In this evaluation, after the alignment stage using MUSCLE, the similarity matrices and the Eigenmaps are calculated using the same algorithms used in SpCLUST. The clustering is then computed using one of the following methods.
- The GaussianMixture() function, from the scikit-learn library that is embedded in SpCLUST.
- The algorithm introduced in Figure 1, which uses the paperrune's C++ GMM implementation and labeled "MaxLikelihood".
- The spectral_embedding() function using the Normalized Laplacian matrix also from the scikit-learn library.
- Each one of the three remaining proposed algorithms, that were proposed and detailed in Section 3, and that use the C++ GMM implementation proposed by this research. The method described in Figure 1, where the maximum likelihood is replaced by the best BIC, is labeled "Fast". The one illustrated in Figure 2 is called "MostFreq" and it executes 500 iterations. Finally, the last algorithm outlined in Figure 3 is named "BestBIC". It executes a maximum of 100 iterations but stops earlier if no improvement is detected after 70 consecutive iterations.
As discussed earlier, the computation of the Laplacian Eigenmap is embedded in the spectral_embedding() function. Conversely, for the GaussianMixture() function, the Eigenmap is computed using functions from the NumPy linear algebra library. For the remaining C++ implementations of the GMM, an implementation [11] of Jacobi's Eigen solving algorithm is used. The used datasets will be also clustered using UCLUST and CD-HIT, which are the best competitors to SpCLUST. Since the spectral_embedding() function does not include any method that facilitates the choice of the adequate number of clusters, this number will be set similarly to the number of clusters produced by SpCLUST.
5. EXPERIMENTAL RESULTS
On the basis of study's set of experiments, the presented GMM implementations in Section 2 and the proposed algorithms detailed in Section 3 are evaluated. The datasets are also clustered using UCLUST and CD-HIT for comparison. In order to cover a wide range of similarities, the identity thresholds chosen for UCLUST and CD-HIT ranged between 0.5 and 0.99, with an incrementation step of 0.1 in the [0.5,0.8[ interval and an incrementation step of 0.01 in the interval. For any identity threshold lower than 0.8, CD-HIT failed to cluster the data. For the sake of comparison, only the produced clusterings having a number of clusters close to the ones from SpCLUST were considered.
To evaluate the quality of each clustering, the degree of similarity between the clustering and the reference must be computed using a relevant metric. Many clustering quality metrics are available in the literature [49, 50]. In this work, the Adjusted Rand Index (ARI) was selected to compute the degree of similarity because it only requires the labels and it is able to compare clusterings with different number of clusters. This index computes a similarity measure between two clusterings by considering all pairs of samples and counting pairs that are assigned in the same or different clusters. It ranges from 0 for two completely different clusterings to 1 for two identical ones.
Table 2 displays, for each dataset and each clustering returned from the considered methods, the number of clusters in both generated and reference clusterings and the ARI between them. Note that ARI is omitted in the following three special cases.
- When a clustering consists of only one cluster.
- When the number of clusters, formed of singletons, is greater than half of the number of sequences (most of the sequences are clustered as one sequence per cluster).
- When the labels of adjacent leaves on the phylogenetic tree are very heterogeneous and the resulting clustering does not reflect any correct grouping on the tree.
The clusterings, matching the first special case, will be discussed later according to the properties of the involved dataset. Conversely, those matching the second case are not significant because the sequences belonging to the same dataset are a priori known to be related.
Table 2. External Clustering Validation Using the Adjusted Rand Index
|
HIV |
NADH |
Influenza |
|||||||
|
Nb. Clusters |
ARI |
Nb. Clusters |
ARI |
Nb. Clusters |
ARI |
||||
|
ref. |
gen. |
ref. |
gen. |
ref. |
gen. |
||||
|
SpCLUST |
4 |
5 |
0.777 |
5 |
4 |
0.957 |
4 |
5 |
0.932 |
|
Paperrune's GMM - MaxLikelihood |
6 |
6 |
0.236 |
11 |
8 |
0.838 |
3 |
3 |
0.847 |
|
sklearn.manifold.spectral_embedding () |
7 |
3 |
0.119 |
7 |
4 |
0.694 |
4 |
3 |
0.653 |
|
Fast |
3 |
3 |
0.801 |
4 |
2 |
0.804 |
- |
1 |
- |
|
MostFreq |
2 |
2 |
0.941 |
4 |
2 |
0.841 |
- |
1 |
- |
|
BestBIC |
3 |
3 |
0.828 |
4 |
3 |
0.839 |
2 |
2 |
1 |
|
UCLUST (id 0.5 ) |
- |
78 |
- |
5 |
6 |
0.374 |
- |
1 |
- |
|
UCLUST (id 0.88 ) |
- |
78 |
- |
- |
83 |
- |
- |
1 |
- |
|
UCLUST (id ) |
- |
78 |
- |
- |
- |
2 |
2 |
1 |
|
|
UCLUST (id ) |
- |
78 |
- |
- |
97 |
- |
3 |
3 |
1 |
|
CD-HIT (id 0.91) |
- |
66 |
- |
- |
90 |
|
- |
1 |
- |
|
CD-HIT (id 0.92) |
- |
69 |
- |
- |
92 |
- |
1 |
2 |
- |
|
CD-HIT (id 0.93-94) |
- |
- |
- |
94-95 |
- |
2 |
2 |
1 |
|
|
CD-HIT (id ) |
- |
- |
- |
- |
3 |
3 |
1 |
||
The MostFreq algorithm scored the best ARI in the case of clustering the HIV set of sequences. The BestBIC version obtained the second rank, followed by the Fast algorithm. As expected, when the number of clusters in the reference clustering and the generated clustering match, the latter obtains a good score. On the other hand, failing to produce the same number of clusters as the reference clustering, might penalize the score of the generated clustering.
For example, SpCLUST produced one more cluster than the reference clustering, and spectral_embedding() produced three clusters less than the reference because in the reference clustering non-adjacent clusters on the tree were not merged. Finally, UCLUST and CD-HIT both failed to cluster this set, although its sequences show a minimum similarity of 86% (cf. Table 1). Indeed, CD-HIT produced 5 clusters when the similarity parameter was set to 0.8, but these clusters do not reflect any meaningful grouping and scored the lowest ARI.
SpCLUST scored the highest ARI values for the NADH dataset, followed by our GMM implementation with the MostFreq and BestBIC approaches respectively. The MostFreq and Fast approaches produced two highly similar clusterings as indicated by their close ARI scores and the same number of clusters. The MaxLikelihood approach detected the largest number of accurate clusters, while MostFreq produced the most accurate clustering among our GMM implementations. As for the HIV set, UCLUST and CD-HIT both failed to cluster this divergent dataset. Although, UCLUST returned a reasonable number of 6 clusters when the identity parameter was set to 0.5, this clustering earned a very low ARI when compared to the other approaches.
The "Fast" method is similar to the one used in SpCLUST, except for the K-Means implementation and the random number generator, which leads to small differences in the results. For the NADH dataset, the seed used in SpCLUST resulted in a better ARI score than BestBIC even though the opposite was expected. This case might occur if the seed of the Fast algorithm is not part of the ones considered in the BestBIC. This situation can be corrected by increasing the set of possible seeds in the BestBIC approach (just 100 different seeds are considered by default). Indeed, three additional experiments using the BestBIC algorithm and involving seeds from outside the scope of the initial experiment, scored an ARI of 0.957 similarly to SpCLUST.
In the Influenza nucleoprotein dataset where the sequences are highly similar, BestBIC scored a perfect ARI similarly to UCLUST and CD-HIT. UCLUST and CDHIT produced equally accurate clusterings, consisting of 3 clusters when the range of identity thresholds was set to 0.95 or higher. However, the BestBIC approach produced a more balanced clustering consisting of 2 clusters, which is similar to the one produced by UCLUST and CD-HIT for a range of thresholds lower than 0.95. Fast and MostFreq, for their parts, produced only a single cluster. This result is not absurd because the sequences in this dataset are considered very similar for a tool that targets clustering potentially divergent datasets. Applying UCLUST and CD-HIT on this dataset, with identities inferior to 0.88 and 0.91 respectively, a single cluster was also produced.
As shown in the previous experiments, traditional tools failed to cluster divergent sequences, while GMM-based approaches have been successful. For instance, even though CD-HIT produced a reasonable number of clusters for the HIV dataset (5 with an identity threshold of 0.8 ), each cluster seems to contain random sequences with no logical grouping and thus, a reference clustering could not be deduced to calculate the ARI. Conversely, despite the fact that UCLUST and CD-HIT succeeded in clustering very similar sequences, similar to that of the Influenza nucleoprotein set, BestBIC also produced a good quality clustering. Therefore, GMM approaches can be considered in most cases regardless of the dataset's degree of similarity.
In terms of current research, the GMM implementation with the BestBIC algorithm obtained the highest average Adjusted Rand Index for the clustering of the three considered datasets, equal to 0.889. It was followed by SpCLUST (using GaussianMixture() from Python's scikit-learn library) and paperrune's GMM implementation with the MaxLikelihood algorithm that scored an average ARI equal to 0.888 and 0.640 respectively. Therefore, on average, the BestBIC approach outperforms the other evaluated tools, in terms of clustering quality, on the chosen datasets. It also competes with the traditional tools when applied on highly similar sequences. For all these reasons, this algorithm was adopted in the next sets of experiments.
After evaluating the quality of the produced clusterings with the three new approaches, a performance comparison between them and SpCLUST was conducted. The tests were applied to the datasets introduced in this article and the dataset of 1049 sequences used in [4] to profile SpCLUST. This experiment was run three times over a machine equipped with an i7-6700 3.4GHz processor. Table 3 shows the best recorded execution times (among the three runs) for clustering the four datasets with the four GMM implementations, which include the computation time of the Eigenmap.
Table 3. Clustering Time Using the Different GMM Implementations and Algorithms
|
|
GaussianMixture () |
Fast |
MostFreq |
BestBIC |
|
HIV |
||||
|
NADH |
||||
|
Influenza |
||||
|
1049 Sequences |
The Fast approach achieved up to 42x speedup when compared to the GaussianMixture() function from Python's scikit-learn library, on the large dataset of 1049 sequences. MostFreq and BestBIC also recorded impressive speedups with this dataset when compared to the GaussianMixture() function. Moreover, the Fast approach achieved higher speed-ups when applied on the three smaller datasets, while the the GaussianMixture() function performed closely to the most complex approach, that is the MostFreq. Therefore, it can be concluded that the proposed algorithms using our C++ GMM implementation outperform scikit-learn's GMM implementation.
6. CONCLUSION
The current research has proposed four GMM-based algorithms, for enhancing the accuracy and the performance of the intervention-free spectral clustering technique for both highly similar and divergent biological sequences. The implementation of these algorithms presents major performance enhancements when compared to SpCLUST. It relies on new C++ implementations of the Gaussian Mixture Model (GMM). The use of these GMM implementations greatly enhances the performance of this technique when compared with the previously used Python GMM implementation. A performance comparison for the clustering phase, between SpCLUST and the implementation of the new algorithms, shows a speed-up ranging from 27x to 42x.
A comparative study, between the proposed algorithms, SpCLUST [4], UCLUST [20], and CD-HIT [19], was conducted over three different datasets of real genomic and protein sequences. In contrast with most of the state-of-the-art tools, the spectral clustering technique aims for an intervention-free and a reasonably fast clustering of datasets, regardless of their level of similarity. Although this technique is not yet expected to compete with traditional tools speed-wise and scalability-wise, the experiments revealed that the proposed algorithms produce competitive clusterings for both highly similar and highly divergent datasets. The validation of the obtained results was based on a novel algorithm for selecting the reference clustering and on a carefully selected external clustering validation index, that is the Adjusted Rand Index.
6.1. Future Directions
Possible future extensions to this work include exploiting more clustering techniques in the biological sequences clustering field, including deep learning ones. Further performance improvement is possible by implementing a parallel computation algorithm for the Eigenmap calculation and the GMM. Finally, defining a novel algorithm for calculating the pairwise similarities without the need for aligned sequences, can further enhance the speed and the scalability of the spectral clustering approach.
Conflict of Interest
The author of the manuscript has no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
Data Availability Statement
The data associated with this study will be provided by the corresponding author upon request.
FUNDING DETAILS
No funding has been received for this research.
Bibliography
- Li Y, Xia F, Wang Y, Yan C, Jia A, Zhang Y. Characterization of a highly divergent Sugarcane mosaic virus from Canna indica L. by deep sequencing. BMC Microbiol. 2019;19:1–8. https://doi.org/10.1186/s12866-019-1636-y
- Pentney W, Meila M. Spectral clustering of biological sequence data. Paper presented at: The AAAI Conference on Artificial Intelligence; July 9–13, 2005; Pittsburgh, Pennsylvania. https://cdn.aaai.org/AAAI/2005/AAAI05-133.pdf. Accessed December 13, 2023.
- Paccanaro A, Casbon JA, Saqi MAS. Spectral clustering of protein sequences. Nucleic Acids Res. 2006;34(5):1571–1580. https://doi.org/10.1093/nar/gkj515
- Matar J, El Khoury H, Charr JC, Guyeux C, Chrétien S. SpCLUST: towards a fast and reliable clustering for potentially divergent biological sequences. Comput Biol Med. 2019;114:e103439. https://doi.org/10.1016/j.compbiomed.2019.103439
- Bocklet T, Maier A, Bauer JG, Burkhardt F, Noth E. Age and gender recognition for telephone applications based on gmm supervectors and support vector machines. Paper presented at: IEEE International Conference on Acoustics, Speech and Signal Processing; March 31–April 4, 2008; Las Vegas, USA. https://doi.org/10.1109/ICASSP.2008.4517932
- Genovese M, Napoli E. ASIC and FPGA implementation of the gaussian mixture model algorithm for real-time segmentation of high definition video. IEEE Transac Very Large Scale Integ Syst. 2013;22(3):537–547. https://doi.org/10.1109/TVLSI.2013.2249295
- McLachlan GJ, Peel D. Finite Mixture Models. John Wiley & Sons; 2004.
- Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B Methodol. 1977;39(1):1–22. https://doi.org/10.1111/j.2517-6161.1977.tb01600.x
- Wu CFJ. On the convergence properties of the EM algorithm. Ann Stat. 1983:95–103.
- McLachlan GJ, Krishnan T. The EM Algorithm and Extensions. John Wiley & Sons; 2007.
- Chretien S, Hero AO. Acceleration of the EM algorithm via proximal point iterations. Paper presented at: IEEE International Symposium on Information Theory (Cat. No. 98CH36252); Auguzt 16–21, 1998; Cambridge, USA. https://doi.org/10.1109/ISIT.1998.709049
- Chretien S, Hero AOIII. Kullback proximal algorithms for maximum-likelihood estimation. IEEE Trans Info Theo. 2000;46(5):1800–1810. https://doi.org/10.1109/18.857792
- Celeux G, Chretien S, Forbes F, Mkhadri A. A component-wise EM algorithm for mixtures. J Comput Graph Stat. 2012;10:697–712. https://doi.org/10.1198/106186001317243403
- Biernacki C, Castellan G, Chretien S, Guedj B, Vandewalle V. Pitfalls in mixtures from the clustering angle. Paper presented at: Working Group on Model-Based Clustering Summer Session, July17–23, 2016; Paris. https://hal.science/hal-01419755/. Accessed December 13, 2023.
- Biernacki C, Chretien S. Degeneracy in the maximum likelihood estimation of univariate Gaussian mixtures with EM. Stat Probabil Lett. 2003;61:373–382. https://doi.org/10.1016/S0167-7152(02)00396-6
- Shi M, Bermak A. An efficient digital VLSI implementation of Gaussian mixture models-based classifier. IEEE Trans Very Large Scale Integ Syst. 2006;14(9):962–974. https://doi.org/10.1109/TVLSI.2006.884048
- Kulis B, Jordan MI. Revisiting k-means: new algorithms via Bayesian nonparametrics. arXiv Preprint. earXiv:1111.0352. 2011. https://doi.org/10.48550/arXiv.1111.0352
- Cheng Y. Mean shift, mode seeking, and clustering. IEEE Trans Pattern Anal Machine Intell. 1995;17(8):790–799. https://doi.org/10.1109/34.400568
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. https://doi.org/10.1093/bioinformatics/btl158
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. https://doi.org/10.1093/bioinformatics/btq461
- Ghodsi M, Liu B, Pop M. DNACLUST: accurate and efficient clustering of phylogenetic marker genes. BMC Bioinfo. 2011;12:e271. https://www.doi.org/10.1186/1471-2105-12-271
- Matias Rodrigues JF, von Mering C. HPC-CLUST: distributed hierarchical clustering for large sets of nucleotide sequences. Bioinformatics. 2013;30(2):287–288. https://doi.org/10.1093/bioinformatics/btt657
- Jiang L, Dong Y, Chen N, Chen T. DACE: a scalable DP-means algorithm for clustering extremely large sequence data. Bioinformatics. 2016;33:834–842. https://doi.org/10.1093/bioinformatics/btw722
- Bruneau M, Mottet T, Moulin S, et al. A clustering package for nucleotide sequences using Laplacian Eigenmaps and Gaussian mixture model. Comput Biol Med. 2018;93:66–74. https://doi.org/10.1016/j.compbiomed.2017.12.003
- Larranaga P, Calvo B, Santana R, et al. Machine learning in bioinformatics. Brief Bioinfo. 2006;7:86–112. https://doi.org/10.1093/bib/bbk007
- Reynolds D. Gaussian mixture models. In: Li SZ, Jain AK, eds. Encyclopedia of Biometrics. Springer; 2009; 827–832.
- Vrieze SI. Model selection and psychological theory: a discussion of the differences between the Akaike information criterion (AIC) and the Bayesian information criterion (BIC). Psychol Methods. 2012;17(2):e228. https://doi.org/10.1037/a0027127
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
- Substitution matrix. Wikipedia. https://en.wikipedia.org/wiki/Substitution_matrix. Accessed September 27, 2018.
- Waterman MS. Efficient sequence alignment algorithms. J Theo Biol. 1984;108(3):333–337. https://doi.org/10.1016/S0022-5193(84)80037-5
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecul Biol Evol. 2013;30(4):772–780. https://doi.org/10.1093/molbev/mst010
- Wright ES. DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinfo. 2015;16:e322. https://doi.org/10.1186/s12859-015-0749-z
- Larking MA, Blackshields G, Brown NP, et al. ClustalW and ClustalX version 2. Bioinformatics. 2007;23(21):2947–2948. https://doi.org/10.1093/bioinformatics/btm404
- Von Luxburg U. A tutorial on spectral clustering. Stat Comput. 2007;17:395–416. https://doi.org/10.1007/s11222-007-9033-z
- Langone R, Alzate C, Suykens JAK. Modularity-based model selection for kernel spectral clustering. Paper presented at: International Joint Conference on Neural Networks; July 31–August 5, 2011; San Jose, USA. https://doi.org/10.1109/IJCNN.2011.6033449. Accessed December 13, 2023.
- Saade A, Krzakala F, Zdeborová L. Spectral clustering of graphs with the bethe hessian. Paper presented at: The 27th International Conference on Neural Information Processing Systems; December 8–13, 2014; Cambridge, United States. https://dl.acm.org/doi/proceedings/10.5555/2968826. Accessed December 13, 2023.
- Dall'Amico L, Couillet R, Tremblay N. Optimized deformed laplacian for spectrum-based community detection in sparse heterogeneous graphs. Paper presented at: 33rd Conference on Neural Information Processing Systems (NeurIPS 2019); December 8–14, 2019; Vancouver, Canada.
- Dall'Amico L, Couillet R, Tremblay N. Revisiting the Bethe-Hessian: improved community detection in sparse heterogeneous graphs. Paper presented at: 33rd Conference on Neural Information Processing Systems, December, 2019; Vancouver, Canada. https://inria.hal.science/hal-02429525/. Accessed December 13, 2023.
- Scikit Learn. Gaussian mixture. https://scikit-learn.org/stable/modules/generated/sklearn.mixture.GaussianMixture.html. Accessed December 13, 2023.
- Scikit Learn. Spectral_embedding. https://scikit-learn.org/stable/modules/generated/sklearn.manifold.spectral_embedding.html. Accessed December 27, 2019.
- Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830.
- GitHub. Paperrune/GMM: C++ implementation of gaussian mixture model. https://github.com/paperrune/GMM. Accessed December 2, 2019.
- GitHub. A gaussian mixture model library featuring BIC and AIC. https://github.com/johnymatar/GMM. Accessed October 10, 2021.
- Raghavan VV, Gudivada VN, Govindaraju V, Rao CR. Cognitive computing: Theory and applications. Elsevier; 2016.
- Campanella JJ, Bitincka L, Smalley J. MatGAT: an application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinfo. 2003;4:1–4. https://doi.org/10.1186/1471-2105-4-29
- Myers EW, Miller W. Optimal alignments in linear space. Bioinformatics. 1988;4(1):11–17. https://doi.org/10.1093/bioinformatics/4.1.11
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. https://doi.org/10.1093/sysbio/syq010
- Lefort V, Longueville JE, Gascuel O. SMS: smart model selection in PhyML. Molecul Biol Evol. 2017;34(9):2422–2424. https://doi.org/10.1093/molbev/msx149
- Wagner S, Wagner D. Comparing clusterings: an overview. Institute for Theoretical Computer Science (ITI). https://publikationen.bibliothek.kit.edu/1000011477. Accessed October 10, 2022.
- Guyeux C, Chrétien S, Tayeh GB, Demerjian J, Bahi J. Introducing and comparing recent clustering methods for massive data management in the internet of things. J Sens Actuator Netw. 2019;8:e56. https://doi.org/10.3390/jsan8040056
1 Having an intra-cluster similarity of 85% or lower [1].
2 https://github.com/johnymatar/SpCLUST-V2/tree/master/src/code
3 which uses standard libraries and does not require any additional software to work.
4 whose methods are inspired from Python's GaussianMixture.
5 Formed by the most significant Eigenvectors computed from the affinity matrix.
6The rand() function is not unified in C++ and, when used under different operating systems, it might generate different output numbers for a same input seed.
7https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html
8https://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi
9http://www.atgc-montpellier.fr/presto/
10https://github.com/johnymatar/SpCLUST-V2/tree/master/src/datasets