
| Review | Open Access |
|---|
A Structure-Based Computational Vaccine Strategy for the Emerging Isfahan Virus: In-Silico Vaccine Designing |

|
|---|
![]() Sajjad Ahmad1, Sidra Amin2, Muhammad Rahiyab1, Rooh Ullah3, Arshad Iqbal1*, Syed Shujait Ali1, Liaqat Ali1, Salman Khan1, Hyat Khan4, and Zahid Hussain1
Sajjad Ahmad1, Sidra Amin2, Muhammad Rahiyab1, Rooh Ullah3, Arshad Iqbal1*, Syed Shujait Ali1, Liaqat Ali1, Salman Khan1, Hyat Khan4, and Zahid Hussain1
1Center for Biotechnology and Microbiology, University of Swat, Pakistan
2Ever Green School and College, Swat, Pakistan
3Center for Animal Sciences and Fisheries, University of Swat, Pakistan
4Department of Genomics, Phenomics and Bioinformatics, North Dakota State University, USA
Isfahan virus (ISFV) is a recently discovered zoonotic disorder that causes painful and severe neurological effects in humans. The virus was initially identified after an epidemic in horses and cattle in 1916, and was later isolated from cattle in Richmond, Indiana in 1925. Currently, no effective vaccine is available for ISFV virus. Therefore, the current work aimed to generate a multi-epitope-based vaccine (MEV) candidate targeting Isfahan virus glycoprotein and large polymerase protein. From these proteins, nine epitopes (three T-cell and B-cell epitopes) were finally designated based on their non-allergenic, non-toxic, and antigenic properties. The selected epitopes, together by suitable adjuvant, enabled the development of an antigenic ISFV-MEV candidate free from allergic reactions. Computational modeling showed that the designed MEV binding strongly interacted with human TLR-3 receptors. Furthermore, immune simulations research established that the ISFV-MEV candidate has the potential to elicit robust immune system reactions in humans. Overall, the outcomes of these in silico studies are promising; however, a subsequent in vivo validation is recommended to confirm its potential as a future vaccine candidate
1. INTRODUCTION
The genus Vesiculovirus, belonging to the family Rhabdoviridae, is spread to sensitive vertebrates hosts via arthropod vectors, primarily sandflies [1]. This genus comprises several medically and veterinary relevant viruses, including as the first species of vesicular stomatitis virus (VSV) Chandipura virus, Maraba virus, Piry virus, and Isfahan virus (ISFV) [2]. ISFV was first identified Phelbotomus papatasi sandflies collected in the Dormian region of Iran in 1975, distinguishing it visibly from the earlier discovery of VSV in livestock in 1916 and 1925 [3, 4]. ISFV is endemic in parts of Asia, particularly in Iran, Turkey, and the republics of Central Asia and may infect both humans and domestic animals, and transmission to susceptible hosts is carried out by biting insects, especially sandfly bites. Although human infections are usually mild or asymptomatic, the virus’s maintenance in insect and rodent populations suggests potential for zoonotic spill-over, underscoring its epidemiological relevance [5-7].
The VSV, historically recognized after the livestock outbreaks in 1916, serves as a prototype for understanding ISFV pathogenesis [8]. In animals, vesicular lesions develop at insect bites sites in animals, including teats, nose, mouth, hooves, and coronary bands [9]. These lesions may cause disability and weight loss as a result of trouble eating, but typically resolve within 7 to 10 days without any major effects [9]. Human infection with VSV can occur via direct contact with sick animals or an accidental contact in a laboratory, leading to asymptomatic cases or mild flu-like symptoms that usually resolve within 5-7 days [7, 10, 11].
The ISFV genomes is composed of non-segmented, single-strand, negative-sense RNA that is about 11 kb long and contains five distinct genes in the sequence 3′-N-P-G-M-L-5′, that translate the glycoprotein (G), nucleocapsid (N), matrix (M), phosphoprotein (P), and polymerase (L) proteins, respectively [12, 13]. Morphologically and pathologically, Isfahan virus (ISFV) is identical to vesicular stomatitis virus (VSV), which has a characteristic bullet-shaped particle (65-180nm) with a ribonucleoprotein core enclosed in a lipid envelope obtained from the host plasma membrane [14]. The N protein remains the extreme prevalent virus-related protein in sick cells and is critical for nucleocapsid formation, viral translation and replication [15].
These two primary strains of the VSV serotypes are New Jersey (VSV-NJ) and Indiana (VSV-IN), which are physically and clinically similar but elicit different neutralizing antibodies [16]. Although ISFV and VSV, produce unique neutralizing antibodies in infected animals 751 VSV-NJ- , outbreaks were recorded in the US between 2004 and 2006, comparative studies enhance understanding of antigenic diversity among Vesiculovirus species [17, 18]. Other member of this genus also includes a number of closely related viral species, including the Piry virus, Maraba Virus, Isfahan Virus and Chandipura viruses [19]. While Chandipura viruse has been linked to encephalistis and influenza-like infections in humans. ISFV primarily circulates among sandflies, rodents, and occasionally humans, without causing disease in domestic animals [18-20].
Experimental studies suggest that ISFV and VSV can be transmitted vertically, supporting the role of insects as primary reservoirs and vectors, which allow the virus to spread and persist in insect populations without the requirement for an intermediate host [21, 22]. VSV infection in humans is very rare, however it can happen when doctors, veterinarians, and animal workers come into close contact with sick cattle and through unintentional exposure to laboratory staff [11]. The mucous membranes of the skin, nose, mouth, and eyes are the most often affected human organs, however there is serological evidence that several vesiculo viruses may be directly transferred to people by insect bites [23]. Human VSV infection can either be asymptomatic or can cause symptoms of the disease, such as myalgia, headaches, and fever, which go away in 3-5 days without any problems [24].
Presently, no specific antiviral agent medications or vaccines exist for ISFV, highlighting is an urgent need for proactive vaccine research other than reactive outbreak control. Computational (Immunoinformatics) vaccine design offers a potential way to advance the creation of reliable and secure vaccinations against recently emerging viral diseases like the Isfahan virus. Recent advances in immunoinformatics allow for the prediction of highly immunogenic epitopes proficient of eliciting strong cellular and humoral immune responses and enable integration of structural modeling, molecular docking, and immune-simulation tools to predict vaccine behavior prior to experimental validation. Structure-based computational vaccine design has successfully been applied to several emerging pathogens, including Zika, Nipah, and Chandipura viruses [25-27].
Using this approach, the present study designed a computational multi-epitope vaccine incorporating Glycoprotein and Large polymerase protein epitopes with L7/L12 as an adjuvant added further to the N-terminal of epitopes and linker by specific sequence for elevated immunogenicity [28]. The researchers first selected their target proteins to predict the potential B-cell and Cytotoxic T-cells and Helper T-cells epitopes. The toxicity test composed with the allergenicity and antigenicity evaluation of the epitopes was performed. The epitope sequences for the vaccine design followed the order established with the better linkers.
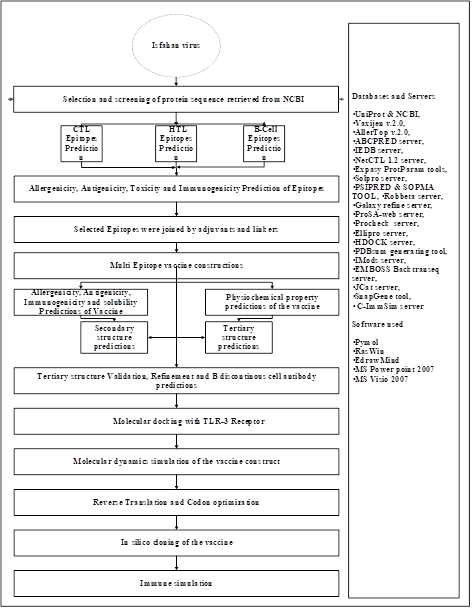
Figure 1. Graphically Overview of the Multi-epitope Vaccine (MEV) Design Strategy Using Both Non-structural and Structural Proteins of the Isfahan Virus
The research team assessed various physical characteristics along with allergic reactions and antigenic features of the design vaccine. The secondary and three-dimensional model were predicted and refined, followed by molecular docking with TLR-3. Molecular dynamics (MD) simulation was conducted to assess the stability of the vaccine receptor complex, integrating computational parameters such as eigenvalues and covariance plots to biological stability. Finally, in-silico cloning and sequence optimization procedures were performed to evaluate the vaccines expression potential. The research methodology displays its framework as displayed in Figure 1.
2. MATERIALS AND METHODS
This research presents a comprehensive in silico immunoinformatics method for the design and assessment of possible epitope-derived subunit vaccines against the Isfahan virus, including the identification of antigenic epitopes, vaccine constructs modeling, and validation of their immunogenic, physicochemical, and structural properties.
2.1. Obtaining Isfahan Viral Proteins for Vaccine Formulations
The peptide sequences of two target proteins of the ISFV, Glycoprotein (G) (Accession ID: CAH17547.1) and large polymerase (L) (Accession ID: CAH17548.1) were received from the NCBI website (https://www.ncbi-nlm.nih.gov//) in the FASTA format. The glycoprotein and large polymerase proteins were identified for vaccine design based on their high antigenicity score and vital roles in viral attachment and replication, respectively, thus representing highly immunogenic and conserved. Other viral proteins such as Nucleocapsid (N), Matrix (M), and Phosphoprotein (P) were excluded because they are primarily internal, show lower antigenicity, have limited surface accessibility and play a major role in immune recognition, reducing their potential to induce strong protective antibody response [29]. The antigenicity of both proteins was confirmed using the VaxiJen v.2.0 website (http://www.ddg-pharmfac.net//vaxijen//VaxiJen//VaxiJen.html/) [30], and the only antigenic proteins were determined for epitope analysis and inclusion in the multi-epitope vaccine formulation [31].
2.2. CTL Epitope Prediction
CTL epitopes targeting these proteins were projected through the NetCTL 1.2 available online website (https://www.google.com//search?q=net-CTL+1.2) [32, 33]. CTL epitope prediction based on three crucial elements, as well as the adhesion of peptides MHC proteasomal its C-terminal cleavage activity. TAP transport accuracy and using a weight matrix as opposed to artificial neural network (ANN), which was used to predict proteasomal C-terminal cleavage and MHC-I binding. Although, Weight Matrix was utilized to expect TAP delivering score. The CTL epitope detection cutoff was set as 0.75 [34].
2.3. HTL Epitope Prediction
Helper T-lymphocyte (HTL) epitopes were forecast via the IEDB MHC-II binding predicated tool (http://tools.iedb.org//mhcii//) [35], with a peptide/chain length set to 15 amino acids by default [[36]]. The analysis identified seven potential epitopes that fix to various HLA class II alleles. In the context of vaccine design, epitopes with lower percentile ranks were prioritized, as these are indicative of stronger binding affinity to the respective HLA molecules.
2.4. B-cell Epitope Prediction
ABCpred predicated online website (http://crdd.osdd..net//raghava/bcepred/) was utilized to predict B-cell epitopes which are essential for the production of defensive antibodies against human [36]. Selection of excellent epitope determined by a high score, with the threshold value set at 0.5
2.5. Construction of Multi-Epitope Vaccine Sequence
After detecting immunogenic epitopes for two proteins, multiple linkers were used to join the predicted epitopes and accomplish the design of the vaccine construct. GPGPG, KK, and AAY, linkers and adjuvant were used to connection these HTL, CTL and B-cell epitopes respectively. These linkers are immunogenic in nature and helpful to increase the immune response in vaccine construction. Furthermore, these linkers prevent epitopes from bending, and maintain them apart from one another. The KK, AAY, GPGPG, and EAAAK linkers were selected because they have been experimentally confirmed in multi-epitope vaccine constructs for improvement of epitope presentation and immune processing. In particularly, AAY enhances proteasomal cleavage and MHC-I presentation, GPGPG improves HTL epitope recognition, KK boosts B-cell exposure, and EAAAK provides rigidity and structural distinction between the adjuvant and epitopes. According to the previous research, E. coli 50S ribosomal subunit can act as potent protective antigens by inducing strong cellular and humoral immune reactions [37, 38]. Therefore, we add adjuvant (50S ribosome protein L7/L12) to the N-terminus portion of the vaccine sequence to improve the stability and immunogenic potential of the generate protein [39]. The L7/L12 adjuvant was chosen because it is a known TLR4 agonist that is known to induce balanced Th1 and Th2 responses. It has been effective in comparable in silico vaccine designs, with high immunogenicity with low allergenic responses and good compatibility with bacterial expression systems. Finally the immunogenicity and allergic reactions of the vaccine design were confirmed utilizing AllerTOP v.2.0 website (https://www.ddg-pharmfac.net//Aller-TOP/), and VaxiJen v.2.0 website (https://www.ddg-pharmfac.net//vaxijen/VaxiJen//VaxiJen.html) [40].
2.6. Domain Identification and Physiochemical Features of the Vaccine Design
Build and normalize some key vaccine construct physiochemical properties with ExPASy ProtParam web tools from (https://web.expasy.org//protparam//). These features included amino acid makeup along with molecular weight and (GRAVY) and theoretical pI and in-vivo and in-vitro half-life instability index. The SOLPRO service served as a platform for protein solubility production through microbe Escherichia coli expression [41].
2.7. Secondary Structure Prediction
Using online PSIPRED service (http://bioinf.cs.ucl.ac.uk//psipred//) is a high-accuracy, publicly accessible online tool identifying secondary (2D) structure of protein from primary amino acid chains [42]. Second, then predict the second structure of the vaccine construct by SOPMA online tool service (https://npsa-prabi.ibcp.fr/cgi-bin/npsa-automat-pl?page=//NPSA//npsa/sopma.htm) [43]. The prediction of secondary structure is important for vaccine design because it provides insights into the folding, stability, and exposure of epitopes, which can influence the vaccine’s immunogenicity and overall structural integrity
2.8. Predictions and validation of Tertiary/3D structures
After verifying that the vaccine design is non allergen and immunogenic in nature, the protein sequence of the established vaccine was submitted to the Robetta service (https://robetta.baker-lab.org//) [44, 45], for tertiary structure modelling structurally. Using automated templates, this website presents a representation of the 3D structure of proteins. The Galaxy refine service (https://galaxy.seoklab.org//cgi-bin/report-REFINE-cgi?key) was utilized to enhance the generated model. They showed the predicted vaccine of 3D structure analyzed the quality of the protein model and then used the ProSa web tools (https://prosa.services.came-sbg.ac.at//prosa.php) to determine the Z score. The protein model’s overall quality is specified through the Z-score [46, 47]. The PROCHECK structural validation service (https://saves.mbi-ucla.edu//) was used to obtain the Ramachandran plot analysis [47, 48]. The preferred, acceptable region, and banned area for amino acid residues. The Ramachandran plot gives the van der Waals radius the side chain is determined by the dihedral angles psi (ψ) and phi (ϕ) of amino acids peptide [49, 50]. This displays the quality of the 3D protein structure and used ERRAT to check for faults in protein structure [51, 52]. The aforementioned websites were utilized to analyze the protein composition based on quality score.
2.9. Prediction of discontinuous/conformational epitopes from B-cell
The modified 3D model of the vaccine was employed to identify and improve the discontinuous epitopes from B cell on the Ellipro website (http://tools-iedb.ore/ellipro/). The Ellipro site provides a score to every predicted epitopes by averaging the Protrusion Index (PI) values over the residues of the planned vaccine epitope [53].
2.10. Molecular docking of the constructed vaccine’s against TLR-3
The negative ssRNA virus Isfahan virus detects recognition through TLR-3 and TLR-8 [54]. In this studies docking was performed against TLR-3 receptor because it is the most relevant receptor involved in recognizing the double-stranded RNA intermediates generated during the replication of negative-sense ssRNA viruses such as the Isfahan virus, TLR-3 serves as a key endosomal pattern recognition receptor that activates antiviral signaling via the TRIF-dependent pathway, inducing the generation of type I interferons and pro-inflammatory cytokines. Although TLR-4, TLR-7, TLR-8, and MHC molecules are also concerned in viral immune recognition, TLR-3 was designated for this study as it represents the primary receptor responsible for detecting the Isfahan virus. Future investigations may include docking analyses with TLR-4, TLR-7, TLR-8, and MHC molecules to explore additional immune recognition pathways. The vaccine docking to the TLR-3 receptor was performed via the HDOCK online service (http://hdock.phys-hust.edu.cn/), which enables analysis of protein interactions evaluation [55, 56]. Chain A of TLR-3 through (PDB ID; 1ZIW) was chosen as the receptor when filling the receptor choice input within HDOCK online service. The Public Database (PDB) file for our vaccine model was provided to the ligand option during docking but all other parameters used the default configurations. The interactions among the vaccine and TLR-3 were further examined via PDBsum tools (http://www.ebi.ac.uk//thornton-srv//databases/pdbsum/). The docking complex PDB file, along with an email address, were uploaded to the server for detailed output on interaction study.
2.11. Molecular Dynamic Interaction between Vaccine and TLR-3
Molecular dynamic is a computer program used to analyze the molecular action of the receptors and epitopes. The MD simulation research was conducted using IMODS website (https://imods.iqfr-csic.es//) to explore the effect of the vaccine, while interacting with the receptor. This website examines the collective movements of protein sequences (torsional space) using NMA inner coordinates and provides exposure to different analysis, such as deformability, eigenvalue, B-factor of linking residues, variance, and covariance. These qualities lead to a correct prediction regarding the stability of the substance [46, 57].
2.12. In-silico Cloning for Vaccine Expression
The EMBOSS Backtranseq online web service (https://ebi.ac-uk//Tools/st/emboss_transeq/) was applied to reverse translate and produce the vaccine gene in a vector [46, 58, 59]. The developed vaccine was cloned in the Escherichia coli (E. coli strain K12) expressing system. We converted vaccine amino acid sequence into a DNA sequence using a Java Codons Adaption Tool server (JCat) ( http://www.jcat.de//). The JCat server’s report for GC content as well as Codon Adaption index (CAI) values indicates the expression system level in E. coli. [46, 60]. The SnapGene tool/ program, two restriction sites, XhoI and NdeI, were implanted to the C and N terminus of the nucleotides to clone the required genes into the E. coli pET-28a (+) expressing vector [46, 59, 61, 62].
2.13. Immune Simulation
The immunogenicity of the designed multi-epitope vaccine was evaluated with the C-Immune simulation, the C-immune tool prediction ( accessed on 25 November 2022 at (https://kraken.iac-rm.cnr.it//C-IMMSIM) which offers predictions of immune system responses to vaccination [63]. The server employs position-specific scoring matrices (PSSM) created via neural network techniques to simulate immunological connections, including B-cell, T-cell, and cytokine feedback. Major Key simulation parameters were defined as follows: three injections of the vaccine with four-week intervals, each containing an antigen dose of 1000 units, and a total simulation duration of 1050 time steps. These parameters were chosen to stimulate a real-life vaccination schedule and to permit evaluation to inspect both primary and secondary immune responses. The antigenicity and immune response profile of the modified peptides were tested under these conditions.
3. RESULTS
3.1. Protein Sequence Retrieval
The Isfahan virus both structural and non-structural proteins, Glycoprotein (GP) (Accession ID: CAH17547.1), and large polymerase protein (LP) (Accession ID: CAH17548.1) amino acid sequence were saved from NCBI in FASTA format, and adjuvant (50S ribosome protein L7/L12) was obtained from UniProt (ID: P9WHE3). Both protein’s antigenicity and allergenicity were evaluated. Table 1 displays the potential allergenic properties of the proteins as determined via AllerTOP 2.0 website and the antigenicity of each protein as evaluated by VaxiJen 2.0 online server with a 0.4 threshold value.
Table 1. Protein’s Sequence of the ISFV Genome Used for Vaccine Design
|
Proteins |
Accession No |
Antigenicity score |
Allergenicity |
|---|---|---|---|
|
Glycoprotein GP |
CAH17547.1 |
0.4964 |
Non allergen |
|
Large polymerase protein LP |
CAH17548.1 |
0.4459 |
Non allergen |
3.2. CTL Epitope Prediction
A total of nine epitopes were forecast via NetCTL 1.2 website and chosen to confirm a higher expected capacity to communicate with multiple MHC class one alleles on the basis of 0.75 values less than 0.4. Nine epitopes were selected based on the class I MHC allele’s ability to bind with them most strongly. Two epitopes were chosen from large polymerase protein and one from glycoprotein. The ability of epitopes to predict toxicity, allergenicity, antigenicity and immunogenicity was further evaluated. Epitopes that caused antigenicity scores below than 0.4 were removed. The three class 1 MHC, epitopes that were chosen for upcoming study are listed in Table 2. The remaining six epitopes were excluded because they did not fulfill the combined selection criteria of strong antigenicity (score > 0.4), non-toxicity, non-allergen, and high MHC class I binding affinity. Only the top three epitopes meeting all parameters were retained to ensure vaccine safety and specificity.
Table 2. Three Class I MHC (CTL) Epitopes Chosen for Further Study
|
Peptide |
Length |
Antigenicity |
Allergenicity |
Toxicity |
Immunogenicity |
|---|---|---|---|---|---|
|
GVDITLVIY |
9 |
1.2950 |
Non allergen |
Non toxin |
0.28108 |
|
QSDLEDLWL |
9 |
0.6579 |
Non allergen |
Non toxin |
0.23448 |
|
VTTCDFRWY |
9 |
0.7588 |
Non allergen |
Non toxin |
0.26325 |
3.3. HTL Epitopes prediction
HTLs produce necessary cytokines that activate humoral immune responses while expressing multiple cytokines essential for eliminating extracellular infections. HTL epitopes serve to stimulate T cell cytokines production that regulates the adaptive immune system. The IEDB website detected 7 HTL epitopes which have 15-mers lengths for the target proteins. Seven MHC class II antigens were evaluated via IEDB server through analysis of HLA DRB1 allelic variants 03:01 and 07:01 and 15:01 together with DRB3 allelic variants 01:01 and 02:02 and DRB4 allele 01:01 besides DRB5 allele 01:01. An evaluation of allergies occurred through the AllerTOP 2.0 database while VaxiJen v.2.0 determined the antigenicity of each sequence. A list of three selected MHC class II epitopes appears in Table 3. The remaining four epitopes were excluded due to lower antigenicity or predicted allergenic properties. The top three HTL epitopes showing the strongest MHC class II binding affinity and non-allergenic behaviour were retained.
Table 3. Three Selected MHC Class II (HTL) Epitopes
|
Alleles |
# |
Start |
End |
Peptide |
Length |
Antigenicity |
Immunogenicity |
|---|---|---|---|---|---|---|---|
|
HLA-DRB1*07:01 |
07 |
55 |
69 |
ALGFAAISVILIIGL |
15 |
1.2317 |
0.5899 |
|
HLA-DRB4*01:01 |
10 |
45 |
59 |
KGWSILNLLVIQRES |
15 |
0.9285 |
0.15927 |
|
HLA-DRB4*01:01 |
10 |
44 |
58 |
QKGWSILNLLVIQRE |
15 |
1.0155 |
0.26165 |
Note. All are non-allergen and non-toxin
3.4. B cells Epitope prediction
ABCpred predicted online service functioned to predict B-cell epitopes which serve as essential components for creating defensive antibodies in humans. We analyzed allergenicity together with antigenicity properties of nine B-cell epitopes extracted from BCpreds server through Allertop v2.0 along with VaxiJen v2.0 web tools. Three different B-cell epitopes displayed high levels of antigenicity without being allergenic or toxic and being immunogenic as noted in Table 4. The selection of top epitopes occurred by rating their scores above 0.5. The B cell epitopes receive presentation in Table 4. The other six predicted epitopes were removed because of either lower antigenicity scores (<0.5), allergenicity, or toxicity predictions. Only three epitopes satisfied all immunogenicity and safety criteria and were used in final vaccine design.
Table 4. The predicted B-cells epitopes are presented in this table.
|
Peptide |
Length |
Antigenicity |
Immunogenicity |
|---|---|---|---|
|
AFVSEGEEVFFGDTGVSKNP |
20 |
0.6397 |
0.26472 |
|
ERVRGRPVPPIWKAKNWTET |
20 |
1.1470 |
0.41355 |
|
VNTTKQLVCWEGQAGGLEGL |
20 |
0.6532 |
0.13712 |
Note. All are non-allergen and non-toxin
3.5. Multi-epitope vaccine (MEV) formulation
The final MEV construct composed of three linear B-cell epitopes, 3 HTL epitopes, and 3 CTL epitopes were combined using the KK, GPGPG, and AAY linkers, as well as demonstrated in Table 5. The vaccine sequence's N-terminal was also linked to the 130 amino acids/peptide long (50S ribosomal L7/L12), adjuvant using an EAAAK linker. The initial vaccine structure as displayed in Table 5 and Figure 2 had 294 amino acids.
Table 5. Epitopes Incorporated into the Final Multi-epitope Vaccine Construct, Selected Based on Immunogenicity, Population Coverage, and Binding Affinity to HLA Alleles
|
Proteins |
CTL epitopes |
HTL epitopes |
B-cells |
|---|---|---|---|
|
GP protein |
VTTCDFRWY |
ALGFAAISVILIIGL |
AFVSEGEEVFFGDTGVSKNP |
|
LP protein |
QSDLEDLWL GVDITLVIY |
KGWSILNLLVIQRES QKGWSILNLLVIQRE |
ERVRGRPVPPIWKAKNWTET VNTTKQLVCWEGQAGGLEGL |
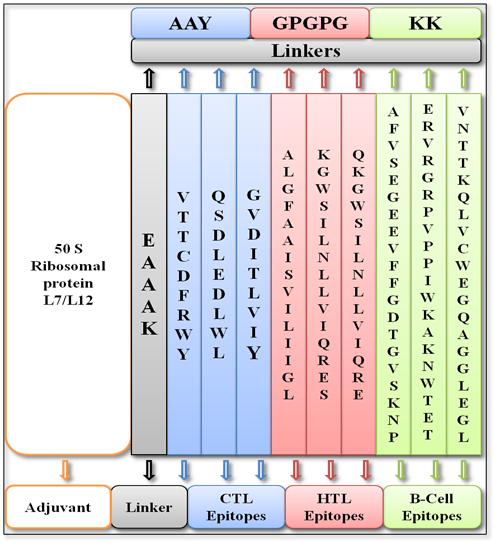
Figure 2. Schematic Representation of the Multi-epitope Vaccine Design, Illustrating Epitope Arrangement and Linker Integration.
3.6 Prediction of Allergenicity, Antigenicity, and Physiochemical Properties of the Vaccine Design
The final vaccine design /construct’s antigenicity and allergic reactions was predicted via VaxiJen and AllerTOP v.2.0 services, our vaccine construct is Non-Allergen, Antigenic, and Immunogenic in nature. The scores for Antigenic and Immunogenic are 0.5266, and 2.9827, respectively. In order to predict the physiochemical characteristic, the ProtParam service was employed. The molecular weight of vaccine is 31189.99. The theoretical PI value at 5.12 indicates acceptable and immunogenic vaccine construction since the measurement of index (II) reached 25.45. The final analysis revealed 98.95 as the aliphatic index combined with 0.037 as the GRAVY (grand average of hydropathicity). The SOLPro server’s prediction of solubility indicated the vaccine natural ability to dissolve with the solubility score of 0.794157. The solubility score suggests that the vaccine designs are extremely soluble while expressed independently in E. coli cells, according to the solubility score. The estimated half-life is 30 hours for mammalian reticulocytes grown in vitro, >20 hours for yeast grown in vivo, >10 hours for Escherichia coli grown in vivo, as shown in Table 6. This solubility score (0.794157) is higher than average values reported for other multi-epitope vaccines (0.65–0.75), suggesting a potentially better solubility and expression efficiency in E. Coli [64, 65].
Table 6. Physiochemical Characteristics of the Designed Vaccine
|
Vaccine construct |
Molecular weight (kDa) |
Theoretical PI |
Half-life in E. coli |
Instability index |
Aliphatic index |
GRAVY
|
Immunogenic score |
|---|---|---|---|---|---|---|---|
|
GP and LP protein |
31189.99
|
5.12 |
>10 hours |
25.45 |
98.95 |
0.037 |
2.9827 |
3.7. Predictions of the Secondary (2D) Structure
The predicted secondary structure of the final vaccine needed SOPMA and PSIPRED server analysis which depended on the protein's estimated amino acid length and predicted (2D) secondary structure. Experimental data on a 294 amino acids sequence revealed 84 amino acids formed coils while alpha-helices needed 123 amino acids to develop and beta turn required 29 amino acids yet beta strands needed only 58 amino acids to form. The overall secondary structure prediction yields 28.57% coil and 41.84% alpha helix with 9.86% beta turn & 19.73% beta strand structures as depicted in Figure 3.
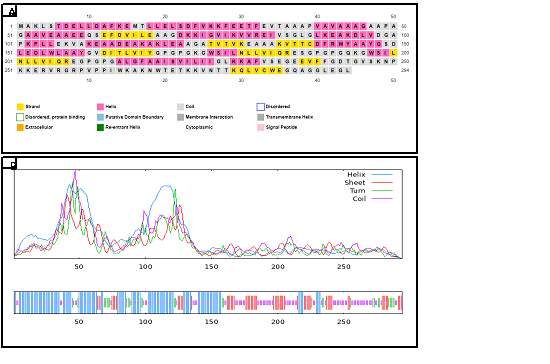
Figure 3. (A) Predicted Secondary (2D) Structure of the Designed Vaccine Construct. (B) The 2D Structure Analysis using the SOPMA Tool
The 2D Structure Analysis using the SOPMA Tool Revealed that the vaccine comprises approximately 41.84% alpha helices, 28.57% random coils, 19.73% extended strands (beta sheets), and 9.86% beta turns.
3.8. 3D Structure Prediction and Enhancement of Vaccine Construct
The prediction of vaccine structure occurred through the Robetta service. Among the five models the first one demonstrated the minimum RMSD value which we selected. The Robetta 3D vaccine structure went through enhancements using Galaxy refine service. The modified five models resulted in selection of model 1 because it demonstrated an optimal GDT-HA score of 0.9804 yet Model 1 also received the best model quality score among the options (Table 7).
Table 7. Summary of Structure Refinement Results from the GalaxyRefine Server
|
Model |
GDT-HA |
RMSD |
MolProbity |
Clash score |
Poor rotamers |
Rama favored |
|---|---|---|---|---|---|---|
|
Initial |
1.0000 |
0.000 |
1.310 |
2.7 |
0.0 |
96.2 |
|
Model 1 |
0.9804 |
0.312 |
1.790 |
11.3 |
0.4 |
96.6 |
|
Model 2 |
0.9872 |
0.305 |
1.819 |
12.2 |
0.4 |
96.6 |
|
Model 3 |
0.9855 |
0.324 |
1.833 |
12.6 |
0.0 |
96.6 |
|
Model 4 |
0.9864 |
0.296 |
1.766 |
10.6 |
0.9 |
96.6 |
|
Model 5 |
0.9923 |
0.280 |
1.838 |
11.7 |
0.4 |
96.2 |
Among the predicted models, Model 1 was identified as the most reliable based on multiple quality assessment parameters, including GDT-HA score, RMSD, MolProbity score, clash score, percentage of poor rotamers, and the proportion of residues in favored regions of the Ramachandran plot.
Greater GDT-HA values suggest a higher quality mode. More stability in our model is shown by a lower RMSD score [59, 62, 66, 67], it utilizes method to determine the space between atoms. The strongest resemblance is indicated by a score is greater than 0.9. The RMSD score in our model is determined the spacing among atoms, a smaller RMSD value denotes more stability. The MolProbity score serves as an indicator for the crystallographic resolution. The RMSD of our model was 0.312. A lower MolProbity level indicates more minor errors [68]. The MolProbity score of Model 1 (1.790) exceeds the value of the original model calculation. The poor rotamers score indicates the percentage of residues that cannot properly rotate their side chains while the Clashes score identifies the number of unwanted atomic distances [69]. The superior structure for the protein emerges from Model 1 because it demonstrates both weak rotamers scores of 11.3 and 0.4 and clash scores of 11.3. The value that Rama favors improves the model quality. Rama is 96.6 percent favored 1n Model 1. The vaccine’s initial and modified 3D models are shown in Figure 4. The reliability multi-epitope vaccine's (MEV) of original and updated models
Table 8. Multiple-epitope Vaccine’s 3D Model Predicted Continuous B-cell Epitopes
|
Start |
End |
Peptide |
No of residues |
Score |
|---|---|---|---|---|
|
247 |
284 |
SKNPKKERVRGRPVPPIWKAKNWTETKKVNTTKQLVCW |
38 |
0.776 |
|
85 |
103 |
VSGLGLKEAKDLVDGAPKP |
19 |
0.762 |
|
1 |
16 |
MAKLSTDELLDAFKEM |
16 |
0.714 |
|
114 |
130 |
DEAKAKLEAAGATVTVK |
17 |
0.679 |
|
229 |
239 |
KKAFVSEGEEV |
11 |
0.677 |
|
24 |
37 |
FVKKFEETFEVTAA |
14 |
0.65 |
|
157 |
161 |
AAYGV |
5 |
0.63 |
|
289 |
294 |
GGLEGL |
6 |
0.572 |
|
209 |
213 |
GPGPG |
5 |
0.532 |
|
58 |
61 |
EEQS |
4 |
0.522 |
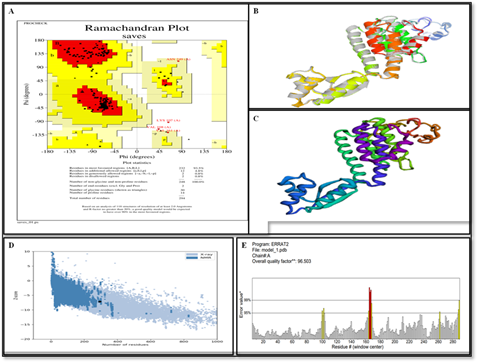
Figure 4. (A) Ramachandran Plot Illustrating the Stereochemical Quality of the Vaccine Model. (B) The Initial 3D Structure of the Vaccine Construct using the Robetta Server. (C) Refined 3D Structure of the Vaccine Model Obtained After Optimization with the GalaxyRefine Websites. (D) ProSA-web Z-score Plot Assessing the Overall Quality of the Refined Model. (E) ERRAT Quality Factor Plot Indicating the Reliability of the Predicted Structure Based on Non-bonded Atomic Interactions
3.9. Tertiary Structure Verification
The ProSa-web and PROCHECK websites were utilized to confirm the tertiary structure as shown in Figure 4. To predict the overall model quality, ProSa offers the Z-score. The Z-score values between 1.26 to 4.74 [67], demonstrated proper compatibility between threading templates in our improved vaccine model. Good alignments between templates occur when Z-score values exceed 1 (>1) [67, 70]. The obtained Z score of -7.15 represented outstanding model quality in our results. The PROCHECK services provide access to view the Ramachandran chart. The results showed that 93.5% of residues occupied most favored regions and 0.8% occupied generously permitted regions while 0.8% residues fell in not allowed zones together with 4.8% further permitted. The protein structure quality factor reached 96.503 points according to ERRAT assessment. The Ramachandran plot displayed in Figure 4 demonstrated the model had superior operation capabilities.
3.10. Discontinuous B-cell epitopes of prediction
Ten discontinuous B-cell epitopes were forecast via Ellipro service, with values ranging from 0.522 to 0.766 (at threshold of 0.5). The discontinuous B-cell epitopes in Table 08, which include epitopes with sizes ranging from 4 to 38 residues, have a score higher than 0.5. Predictions can be made because the protein structure and folding potential may generate new B-cell epitopes of conformational or discontinuous types. The improved 3D model utilized the Ellipro service to detect future conformational/discontinuous B-cell epitopes. The Ellipro service identified seven novel conformational/discontinuous B-cells epitopes in Table 9 and figure 5 at different length sizes from 5 to 59 residues with epitope values between 0.532 to 0.813.
Table 9. Discontinuous/conformational B-cell Epitopes Predicted by Ellipro Server
|
Residues |
No. of residues |
Score |
|---|---|---|
|
A:M1, A:A2, A:K3, A:L4, A:S5, A:T6, A:D7, A:E8, A:L9 |
9 |
0.813 |
|
A:I66, A:L67, A:E68, A:A69, A:A70, A:G71, A:D72, A:K73, A:K74, A:I75, A:G76, A:V77, A:I78, A:K79, A:V80, A:V81, A:R82, A:E83, A:V85, A:S86, A:G87, A:L88, A:G89, A:L90, A:K91, A:E92, A:A93, A:K94, A:D95, A:L96, A:V97, A:D98, A:G99, A:A100, A:P101, A:K102, A:P103, A:L104, A:A109, A:K110, A:E111, A:D114, A:E115, A:K117, A:A118, A:K119, A:L120, A:E121, A:A122, A:A123, A:G124, A:A125, A:T126, A:V127, A:T128, A:V129, A:K130 |
57 |
0.738 |
|
A:A157, A:A158, A:Y159 |
3 |
0.724 |
|
A:K229, A:K230, A:A231, A:F232, A:V233, A:S234, A:E235, A:G236, A:E237, A:E238, A:V239, A:V246, A:S247, A:K248, A:N249, A:P250, A:K251, A:K252, A:E253, A:R254, A:V255, A:R256, A:G257, A:R258, A:P259, A:V260, A:P261, A:P262, A:I263, A:W264, A:K265, A:A266, A:K267, A:N268, A:W269, A:T270, A:E271, A:T272, A:K273, A:K274, A:V275, A:N276, A:T277, A:T278, A:K279, A:Q280, A:L281, A:V282, A:C283, A:W284, A:G286, A:Q287, A:A288, A:G289, A:G290, A:L291, A:E292, A:G293, A:L294 |
59 |
0.712 |
|
A:F24, A:K26, A:K27, A:F28, A:E29, A:E30, A:T31, A:F32, A:E33, A:V34, A:T35, A:A36, A:A37 |
13 |
0.671 |
|
A:D11, A:A12, A:K14, A:E15, A:M16 |
5 |
0.645 |
|
A:G209, A:P210, A:G211, A:P212, A:G213 |
5 |
0.532 |
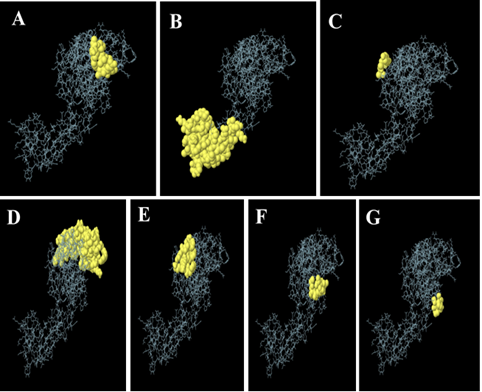
Figure 5. Seven Epitopes for Conformational B-cells. (A) Protein 9-residue Region with a Score of 0.813 (B) Protein 57-residue Region with a Score of 0.738 (C) Protein 3-residue Region with a Score of 0.724 (D) Protein 59-Residue Region with a Score of 0.712 (E) Protein 13-residue Region with a Score of 0.671 (F) Protein 5-residue Region with a Score of 0.645 (G) Protein 5-residue Region with a Score of 0.532
Seven newly predicted conformational B cells epitopes were included in the final enhanced vaccine structure, along with its 3D structure. The yellow portions represent the conformational B-cells epitopes, while the grey sections show the remaining residues
3.11. Molecular Docking of the Designed Vaccine Against TLR-3
The capability of ligand-receptor molecular interactions was assessed through the application of molecular docking in order to determine complex bindings strength and stability. Molecular docking centered on Toll-like receptor 3 because it functions as the vital element to activate immune response and detect infections. The vaccine binds with Chain-A of TLR-3 receptor at the HDOCK server PDB ID: (1ZIW). The vaccine demonstrates strong receptor binding with TLR-3 because of its high affinity but we chose the docking model with the negative energy score of -266.16 [40, 44, 59, 62, 67] (see Table 10). The PDBsum server identified 243 non-bonded contacts and detected 05 salt bridges together with 09 hydrogen bonds during the docked complex interaction analysis. The binding mechanism of the complexes was assessed by PyMOL and revealed through Figure 6.
Table 10. Evaluation of the Top-ranked Refined Docking Complex between the Vaccine Construct and TLR-3, as Predicted by the HDOCK Server
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Docking score | -281.36 | -269.86 | -266.16 | -265.86 | -263.29 | -263.28 | -260.93 | -258.96 | -258.59 | -256.96 |
| Confidence score | 0.9326 | 0.9166 | 0.9108 | 0.9103 | 0.9060 | 0.9060 | 0.9019 | 0.8984 | 0.8977 | 0.8947 |
| Ligand rmsd | 95.02 | 78.90 | 66.59 | 77.16 | 97.04 | 68.75 | 63.20 | 69.07 | 62.47 | 79.92 |
| Interface residues | Model-1 | Model-2 | Model-3 | Model-4 | Model-5 | Model-6 | Model-7 | Model-8 | Model-9 | Model-10 |
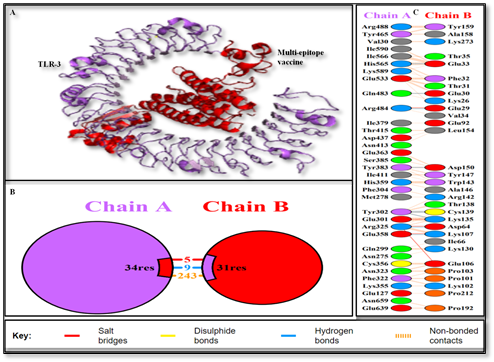
Figure 6. (A) The Protein–protein Docking Complex Illustrates the interaction between the designed Vaccine Construct and TLR-3. (B) and (C) Provide Detailed Views of the Binding Interface Between the Vaccine (Shown in Purple-red) and TLR-3 (Shown in Purple).
The interacting residues from TLR-3 (A chain) and the vaccine (B chain) are highlighted. Several key hydrogen bonds are observed at the interface, suggesting stable and specific interactions within the vaccine-TLR-3 complex.
3.12. Molecular (MD) Simulation of the TLR-3 Docked Complex Vaccine
Users accessed the IMODS websites through the web to perform extensive structural dynamics assessments on the TLR-3 docked complex vaccine construct. The results presented in Figure 7 show the findings, enabled scientists to establish deformability graphs which became the basis for measuring adaptability at each residue position. Scientists expected that upcoming B-factor and NMA testing protocols would undergo modifications. The generated eigenvalue plot presented the relative stiffness data of the model structure. The eigenvalue value tends to decrease when the deformation becomes simpler. The eigenvalue obtained from the results was 5.117716e-05. The analysis shows that the relationship between eigenvalues and normal mode variance operates in direct contrast to one another. The graphical representation of macromolecule section movements appears within a covariance map which shows white areas for uncorrelated regions but displays red regions as correlated while blue areas represent anti correlated sections. The results an elastic network graphs. The hue intensity of the graphic depicts spring hardness where stronger springs appear darker while weaker springs appear lighter.
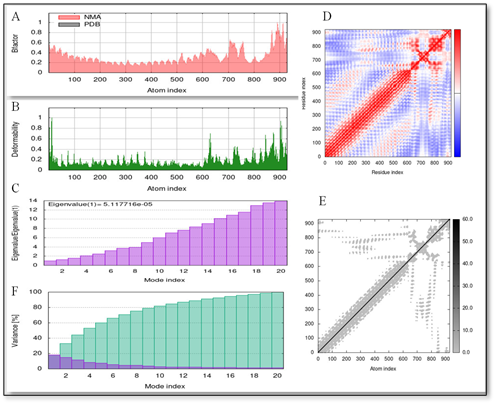
Figure 7. Normal Mode Analysis (NMA) (A) Experiment B-factor, (B) Deformability, (C) Eigenvalue Plot, (D) Co-variance Map, (E) Elastic Network Graph, and (F) variance.
The parameters in Figure 7 collectively demonstrate the vaccine’s mechanical stability and flexibility, supporting its potential for efficient receptor binding
3.13. In silico Cloning for Vaccine Expression
The inverse translation of the vaccine sequence through EMBOSS Backtranseq tool underwent JCat optimization for improved expression from the Java codon adaptation tool. The optimized vaccine sequence exhibited 882 nucleotides with CIA equal to 0.46 and GC content measuring 69.04 %. To achieve uniform plasmid expression in E. coli it is necessary to maintain GC content within 30-70 range. The SnapGene software applied restriction sites NdeI and XhoI into the modified sequence before it was inserted into the pET-28a (+) vector as shown in Figure 8.
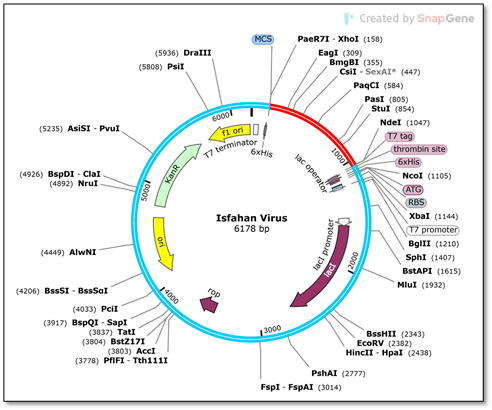
Figure 8. In-silico Cloning of the Vaccine Gene into the pET-28(a) Vector for Expression Optimization
3.14. Immune Simulation
The immune response is intimately coupled with dynamic movements of protein atoms, and molecular dynamics simulations afford a dependable approach to evaluating the structural stability and conformational consistency of the vaccine. The ability of vaccine to stimulate the immune system and its entire immune profile was evaluated using the C-IMMSIM database, that employs automated machine learning algorithms to simulating immune reactions across key anatomical compartments, such as bone marrow, thymus, and lymph nodes. To ensure reproducibility of the predictions, the simulations were repeated three times, under identical input conditions, producing consistent antibody, cytokine, and T-cell response profiles. Figure 09 presents the overall immune response elicited by the vaccine, with highlighting elevated levels of IgM and IgG antibodies. Detailed insights through distinct panels: panel (A) illustrates the number of active B cells, panel (B) depicts the total counts of memory cells, B cells, and immunoglobulins, panel (C) shows the levels of immunoglobulins and antigens categorized according to isotype, and panel (D) represents the CD4⁺ T-helper lymphocyte populations divided into active, resting, anergic, and replicating states. Both helper T lymphocytes (HTL) and cytotoxic T lymphocytes (CTL) established vigorous responses, with extensive increases in IL-2 and IFN-γ production, showing the vaccine’s strong immunostimulatory capacity.
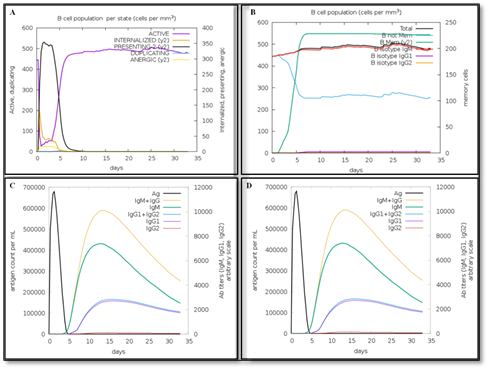
Figure 9. Simulated Immune Responses (A) The number of active B cells. (B) Total number of Memory Cells, Immunoglobulin, and B cells. (C) Immunoglobulins and Antigens. Antibodies are Categorized into Isotype Groups. (D) CD4 T-helper Lymphocytes May be Separated into Active, Resting, Anergic, and Replicating Entity-states.
4. DISCUSSION
ISFV is a global health issue and a repetitive virus for communities that suffer due to a lack of successful protective or treatment strategies [71-73]. Bioinformatics technologies can predict many properties (structural and physiochemical traits) of biomolecular structures like proteins and epitopes and their interactions with targets. One of the benefits of these approaches is their ability to predict rapid, reliable, and efficient basic results [74, 75].
The MEV candidate’s constructions contain both structural and non-structural viral proteins, including glycoprotein and large polymerase protein. These proteins influence immune evasion, viral binding and assembly, viral binding and assembly, INF and NF-B promoter inhibition, JAK-STAT signaling, and viral transcription [76-79]. MEV candidates can elicit stronger reactions than a single protein. Particularly, adjuvants that stimulate humoral and cell-mediated responses can be used to increase the immunogenicity of vaccines. Clinical evaluations of subunit vaccines have demonstrated their capacity to stimulate the antibody response [80]. The rapid collection of extensive genomic and proteomic data on infections, like Isfahan virus, is beneficial for developing of efficient vaccines based on epitopes that can control and eliminate the illnesses carried upon various pathogens.
This research project applied Computational algorithms to predict three types of epitopes including linear B-cell along with HTL and CTL. The selection of CTL and HTL epitopes began because they connect with sufficient HLA alleles for future examination. The selected CTL epitope together with HTL epitope and linear B cell epitope demonstrates both effective antigenicity and determination while also showing non-toxicity and non-allergenicity to vaccine candidates. Several immunoinformatics methods performed the analysis to evaluate the epitopes [81]. Several linkers were used to combine epitopes with different pattern types in the essential components of the vaccine. The physicochemical abilities including stability analysis show that AAY, GPGPG and KK links work effectively to combine B-cell linear epitopes CTL and HTL with each other. The vaccine researchers chose to add the 50S ribosomal proteins L7/L12 adjuvant as well as an adjuvant and 3 CTL epitopes, three in HTL epitopes, all 3 B-cell epitopes, one linker EAAAK, two linkers AAY, three linkers GPGPG, and three KK linkers to the original vaccine's 294 amino acids from the beginning of the amino terminus. The rigid EAAAK linker establishes helix structure elements that boost immunogenic properties as noted by [81, 82]. Several elastic and flexible amino acids integrate within the linkers GPGPG, AAY, and KK resulting in avoidance of area separation [82, 83].
A study of vaccine physicochemical properties enabled researchers to simplify experimental methods for vaccine testing and plan for future both in vitro and in vivo assessments. According to the predicted pI value the vaccine maintains an acidic nature with a calculated 5.12 value. A GRAVY score of 0.037 indicates that the vaccine has hydrophobic properties and high solubility according to [81, 83, 84]. Locate the information in the articles by [81, 85]. A lower GRAVY value exists in our vaccine structure. The experimental vaccine longevity for fighting E. coli was 30 hours while yeast required 20 hours and the mammalian strain required 10 hours of protection. Strengthened immune reactions occur because the vaccine requires more repeated treatments [85, 86]. The vaccine showed no difference between its actual half-life and its predicted half-life according to Kadam et al [85]. The vaccine researchers identified its molecular weight at 31.18 kDa. Small amino acids can be efficiently cloned and expressed in biological systems thus vaccines containing these amino acids under 110 kDa molecular weight present strong vaccination potential [86, 87]. Stability at warm temperatures increases based on the vaccination aliphatic index score which surpasses 50 points [88], as the measured aliphatic index reached 98.95. The 25.45 vaccine instability score indicates that the vaccination retains its stability attributes [88]. The Kadam et al [85], the stability ratio of 38 in the previous research study reflects weaker stability compared to our vaccine formulation. The publication suggests that proteins indicated as stable have instability indexes below 40 [89]. The SOLPro services computed an excellent solubility score of 0.794157 to assess the antigen [64, 65]. The evaluation from VaxiJen v.20 and IEDB servers showed outstanding results for both immunogenicity and antigenicity properties of our vaccine design which obtained scores of 2.9827 and 0.5266 respectively. The AllerTOP v.2.0 website determined, that the designed vaccine did not contain allergenic properties. Our designed vaccine achieved all target components of its physiochemical characteristics. Secondary structures of the complete vaccine were predicted using the analysis tools available at SOPMA and PSIPRED servers. Protein secondary structure prediction required sequence analysis of amino acid substrates. A 294 amino acid sequence produced the different super secondary elements through 84 amino acids for helix formation and 123 amino acids for alpha helices as well as 58 amino acids for beta strands and 29 amino acids for beta turns. The total 2D structure prediction reveals that coils comprise 28.57 percent of the structure while alpha helixes represent 41.84 percent and beta turns stand at 9.86 percent along with beta strands accounting for 19.73 percent. The vaccine showed adequate results because it contained sufficient helices within its predicted secondary structure and hydrogen bond strength [40, 44, 59, 62, 67, 89].
The Robetta server provided tertiary structure identification while GalaxyRefine served to enhance the created vaccine 3D model structure. PROCHECK & ProSa-web databases evaluated the enhanced 3D structure of the vaccine through Z-score Ramachandran plot analysis while ERRAT established an overall quality factor at 96.503. The Z-score of the improved model became -7.15 and the vaccine model displayed optimal Ramachandran plot values 93.5 percent of the time after revision [59, 90]. Our model demonstrates exceptional quality due to the relation between lower z-scores and superior quality of 3D produced vaccine models [87, 89]. We docked the vaccine model through HDOCK service to TLR-3 receptor then analyzed their connections using PDBsum due to TLR3 receptors which best detect Isfahan virus. These receptor interactions were identified. The vaccination showed maximum affinity for the TLR-3 receptor through its detection of 05 salt bridges together with 09 hydrogen bonds and 243 non-bonded contacts. The iMODS website conducted MD simulations to determine stable conditions of TLR-3 vaccine docking complexes [59]. The elastic network between proper atoms with deformability and B- factor along with linking residues atomic properties determine the variance and co-variance of this position. Testing proved the TLR-3 vaccine complex keeps its stability while remaining flexible at the same time. Further the Normal mode analysis (NMA) provided insights into the construct’s dynamic stability. A high eigenvalue suggested strong structural rigidity, while covariance and deformation plots showed limited flexibility within the linker regions, which is desirable feature for maintaining epitope accessibility without compromising the molecular stability of the protein. This shows the MEV can retain its conformational integrity during antigen processing [91].
Immune simulation using C-ImmSim provided additional insights into the vaccine’s potential immunogenicity. The simulation predicted the activation of both humoral and cellular immune responses over repeated antigen exposures. Specifically, elevated levels of immunoglobulins (IgG and IgM) were observed, along with increased populations of helper T-cells (Th1 and Th2), cytotoxic T-cells, and memory B- and T-cells. Key cytokines, including IFN-γ and IL-2, were also produced at high levels, indicating robust cell-mediated immune activation.
The In silico immune simulation also demonstrated primary IgM and secondary IgG responses upon successive antigenic exposures, reflecting strong humoral memory potential. To ensure realistic modeling, the simulation parameters were chosen based on standard vaccination protocols: three injections were administered at intervals corresponding to standard vaccination schedules, and the antigen dosage was set to match the predicted effective epitope concentration. These parameters allowed the simulation to mimic realistic immune responses and provide a meaningful prediction of the vaccine’s immunogenicity. These results suggest that the designed vaccine can induce a strong, long-lasting immune response, supporting its potential efficacy prior to experimental validation.
The vaccine sequence for expression enhancement used the EMBOSS Backtranseq service together with the Java Codon Adaptation Tool (JCat). A result of more than 0.8 may be considered desirable since it indicates matching codon preferences but the optimal CAI score should be 1.0 [84, 92]. The optimized codon found 69.04% in the vaccine derived sequence which had a Codon adaptation index (CIA) of 0.46 and 882 nucleotides [93]. The modified sequence with XhoI and NdeI restriction sites was entered using SnapGene into pET-28(+) vector, suggesting that the vaccine construct can be efficiently expressed in bacterial systems. Studies confirmed that T memory cells released elevated levels of Ig production along with increased levels of IL-2 and IFN-γ while cytotoxic cells numbers also reached high levels [94, 95].
These findings are in strong agreement with recent literature, where similar multi-epitope vaccine constructs against RNA viruses like Ebola, Nipah, and Rift Valley fever virus showed promising immunogenic profiles in silico [40, 44, 59, 62, 67]. Additionally, several genomic and structural similarities between the Isfahan virus and other phleboviruses, suggest that our MEV construct could potentially provide cross-protection against closely related viral strains. This study also shows the first structured attempt to designing a multi-epitope vaccine against ISFV, through alignment to One Health priorities by addressing a neglected vector-borne pathogen with zoonotic potential in sandfly and rodent reservoirs.
Overall, the outcomes obtained in this study highpoint that the designed MEV vaccine establishes a balanced physicochemical, structural, and immunogenic profile, which make it suitable for expression and testing in experimental models [ 94 , 95 ] . Therefore, this vaccine could help as a potential future candidate for preventive immunization against ISFV after further experimental validation. Future studies should focus on in vitro and in vivo evaluations to evaluate immunogenicity, safety, and cross-protection efficiency in animal models . The research presents a new vaccination strategy as a solution for addressing with ISFV condition antigenic characteristics. The predicted vaccine demonstrates immunization potential and has the potential to eliminate the disease based on immunoinformatics methods. Vaccine activity needs to be confirmed through in vitro studies of the immune system [94, 95].
4.1. Limitations
This study is not without its limitations, since experimental validation is lacking despite the comprehensive computational evaluation. Antigenicity, allergenicity, and immunogenicity, as predicted, require in vitro and in vivo confirmation to establish the safety and efficacy of the proposed construct. Furthermore, the limited epidemiological data on ISFV constrains the immediate translational potential of the vaccine. Future work should therefore focus on cloning, expression, and animal model testing to corroborate these theoretical findings.
4.2. Conclusion
The aim of this work was to establish an ISFV multi-epitope vaccine using immunoinformatics techniques as an alternative to conventional vaccine process. The multi-epitope vaccines include CTL, HTL and B-cell epitopes joined via suitable linker sequences provide cell mediated, humoral and innate immunity. It is non-allergenic and demonstrates favorable physiochemical characteristics, high antigenicity, immunogenicity, and stability. Using molecular docking analysis and MD simulation, confirmed strong binding to TLR3. While codon optimization and CAI/GC content analysis indicate efficient production in bacterial hosts. Overall, this multi-epitope vaccine candidate shows promising immunological potential and warrants further in-vitro and in-vivo testing as an ISFV vaccine candidate.
Conflict of Interest
The authors of the manuscript have no financial or non-financial conflict of interest in the subject matter or materials discussed in this manuscript.
Data Availability Statement
All datasets generated and analyzed during this study are included within the article and its supplementary materials. Additional data are available from the corresponding author upon reasonable request.
Funding Details
No funding has been received for this research.
REFERENCES
- Knipe DM, Howley PM, eds. Fields Virology. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007.
- Tesh RB, Travassos da Rosa APA, Travassos da Rosa JFS. Antigenic relationship among rhabdoviruses infecting terrestrial vertebrates. J Gen Virol. 1983;64(1):169-176. https://doi.org/10.1099/0022-1317-64-1-169
- Sia G, Tesh RB, Travassos da Rosa APA, et al. Isolation of the Isfahan virus in Turkmenia. Acta Virol. 1980;24(5):618-620.
- Tesh RB, Travassos da Rosa APA, Travassos da Rosa JFS. Isfahan virus, a new vesiculovirus infecting humans, gerbils, and sandflies in Iran. Am J Trop Med Hyg. 1977;26(2):299-306. https://doi.org/10.4269/ajtmh.1977.26.299
- Wilks CR, House JA. Susceptibility of various animals to the vesiculoviruses Isfahan and Chandipura. Epidemiol Infect. 1986;97(2):359-368. https://doi.org/10.1017/S002217240006544X
- Travassos da Rosa JFS, Tesh RB. Isfahan virus, a new vesiculovirus infecting, gerbils, and sandflies. J Trop Med. 1982;29(1):291-302.
- Nasar F, Palacios G, Gorchakov RV, et al. Recombinant Isfahan virus and vesicular stomatitis virus vaccine vectors provide durable multivalent single-dose protection against lethal alphavirus challenge. J Virol. 2017;91(8):e01729-16. https://doi.org/10.1128/JVI.01729-16
- Cotton WE. The causal agent of vesicular stomatitis proved to be a filter-passing virus. J Am Vet Med Assoc. 1926;69:168-179.
- Letchworth GJ, Rodriguez LL, Del Cbarrera J. Vesicular stomatitis. Vet J. 1999;157(3):239-260. https://doi.org/10.1053/tvjl.1998.0303
- Johnson KM, Vogel JE, Peralta PH. Clinical and serological response to laboratory-acquired human infection by Indiana type vesicular stomatitis virus. Am J Trop Med Hyg. 1966;15(2):244-246. https://doi.org/10.4269/ajtmh.1966.15.244
- Fields BN, Hawkins K. Human infection with the virus of vesicular stomatitis during an epizootic. N Engl J Med. 1967;277(19):989-994. https://doi.org/10.1056/NEJM196711092771901
- Howley PM, Knipe DM, eds. Fields Virology: Emerging Viruses. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2020.
- Ball LA, White CN. Order of transcription of genes of vesicular stomatitis virus. Proc Natl Acad Sci USA. 1976;73(2):442-446. https://doi.org/10.1073/pnas.73.2.442
- Ge P, Tsao J, Schein S, et al. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science. 2010;327(5966):689-693. https://doi.org/10.1126/science.1181766
- Whelan SPJ, Barr JN, Wertz GW. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol. 2004;283:61-119.
- Ferris NP, Nordengrahn A, Hutchings GH, et al. Development and laboratory evaluation of two lateral flow devices for detection of vesicular stomatitis virus in clinical samples. J Virol Methods. 2012;180(1-2):96-100. https://doi.org/10.1016/j.jviromet.2011.12.010
- McCluskey BJ, Peters D, Hannemann R, et al. Vesicular stomatitis outbreak in the southwestern United States, 2012. J Vet Diagn Invest. 2013;25(5):608-613. https://doi.org/10.1177/1040638713497945
- Perez AM, Pauszek SJ, Jimenez D, et al. Spatial and phylogenetic analysis of vesicular stomatitis virus overwintering in the United States. Prev Vet Med. 2010;93(4):258-264. https://doi.org/10.1016/j.prevetmed.2009.11.003
- Tolardo AL, Gómez MM, Fernandes M, et al. Real-time reverse transcriptase polymerase chain reaction for detection and quantification of vesiculovirus. Mem Inst Oswaldo Cruz. 2016;111:385-390. https://doi.org/10.1590/0074-02760150456
- Maia-Farias A, Carvalho JG, Barros-Aragão FGQ, et al. Early and late neuropathological features of meningoencephalitis associated with Maraba virus infection. Braz J Med Biol Res. 2020;53:e9316. https://doi.org/10.1590/1414-431X20208604
- Tosh RB, Chaniotis BN, Johnson KM. Vesicular stomatitis virus Indiana serotype: multiplication in and transmission by experimentally infected phlebotomine sandflies (Lutzomyia trapidoi). Am J Epidemiol. 1971;93(6):491-495. https://doi.org/10.1093/oxfordjournals.aje.a121284
- Ajamma YU, Onchuru TO, Ouso DO, et al. Vertical transmission of naturally occurring bunyamwera and insect-specific flavivirus infections in mosquitoes from islands and mainland shores of Lakes Victoria and Baringo, Kenya. PLoS Negl Trop Dis. 2018;12(11):e0006949. https://doi.org/10.1371/journal.pntd.0006949
- Calisher CH, Gutierrez E, Manzione N, et al. A newly recognized vesiculovirus, Calchaqui virus, and subtypes of Melao and Maguari viruses from Argentina, with serologic evidence for human and equine infection. Am J Trop Med Hyg. 1987;36(1):114-119. https://doi.org/10.4269/ajtmh.1987.36.114
- Jonkers AH. The epizootiology of the vesicular stomatitis viruses: a reappraisal. Am J Epidemiol. 1976;104(3):286-291.
- Shabbir MA, Ahmad I, Haq IU, et al. Immunoinformatics-driven design of a multi-epitope vaccine against Nipah virus. J Genet Eng Biotechnol. 2025;23(2):e100482. https://doi.org/10.1016/j.jgeb.2025.100482
- Hakami MA. Immunoinformatics and structural vaccinology approach to design a multi-epitope vaccine targeting Zika virus. BMC Chem. 2024;18(1):e31.
- Deb D, Srivastava R, Kumar S, et al. Immunoinformatics-based design of a multi-epitope vaccine against Chandipura vesiculovirus. J Cell Biochem. 2022;123(2):322-346.
- Obaidullah AJ, Ahmed I, Khan T, et al. Immunoinformatics-guided design of a multi-epitope vaccine against SARS-CoV-2. RSC Adv. 2021;11(29):18103-18121.
- Vita R, Overton JA, Greenbaum JA, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43(Database issue):D405-D412. https://doi.org/10.1093/nar/gku938
- Haq IU, Ahmad I, Shabbir MA, et al. Computational immunoinformatics approach to design a multi-epitope vaccine against Guanarito virus. World J Biol Biotechnol. 2025;10(1):25-35. https://doi.org/10.33865/wjb.10.1.1473
- Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007;25(5):856-866. https://doi.org/10.1016/j.vaccine.2006.09.032
- Larsen MV, Lundegaard C, Lamberth K, et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:e424. https://doi.org/10.1186/1471-2105-8-424
- Bibi B, Ahmad I, Haq IU, et al. Designing a multi-epitope-based vaccine against rubella virus using immunoinformatics approaches. Microbe. 2025;7:e100323. https://doi.org/10.1016/j.microb.2025.100323
- Ahmad I, Haq IU, Shabbir MA, et al. Development of a multi-epitope subunit vaccine for protection against norovirus infections using computational vaccinology. J Biomol Struct Dyn. 2022;40(7):3098-3109.
- Haq IU, Ahmad I, Shabbir MA, et al. Rational in silico design of a multi-epitope vaccine against human rhinovirus using immune simulation and molecular dynamics approaches. Vacunas. 2025;26:e500427.
- Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):40-48. https://doi.org/10.1002/prot.21078
- Goel D, Bhatnagar R. Intradermal immunization with outer membrane protein 25 protects BALB/c mice from virulent Brucella abortus Mol Immunol. 2012;51(2):159-168. https://doi.org/10.1016/j.molimm.2012.02.126
- Goel D, Rajendran V, Bhatnagar R. Cell-mediated immune response after challenge in Omp25 liposome-immunized mice contributes to protection against virulent Brucella abortus Vaccine. 2013;31(8):1231-1237. https://doi.org/10.1016/j.vaccine.2012.12.043
- Rahiyab M, Ahmad I, Shabbir MA, et al. Design of a new multi-epitope subunit vaccine to combat the EIA virus, targeting Pol, Gag, and Env proteins: in silico technique. Vacunas. 2025;26:e500463. https://doi.org/10.1016/j.vacun.2025.500463
- Khan A, Ahmad I, Haq IU, et al. Designing a highly antigenic multi-epitope subunit vaccine against bovine alphaherpesvirus 2 targeting glycoproteins B and H. Med Omics. 2025;8:e100047.
- Hon J, Marusiak M, Martinek T, et al. SoluProt: prediction of protein solubility. Bioinformatics. 2019;35(9):1462-1464. https://doi.org/10.1093/bioinformatics/bty945
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195-202. https://doi.org/10.1006/jmbi.1999.3091
- Deléage G. ALIGNSEC: viewing protein secondary structure predictions within large multiple sequence alignments. Bioinformatics. 2017;33(24):3991-3992. https://doi.org/10.1093/bioinformatics/btx577
- Hussain I, Ahmad I, Haq IU, et al. Structure-based in silico discovery of thymidine kinase inhibitors targeting goatpox virus. ChemistrySelect. 2025;10(37):e03462. https://doi.org/10.1002/slct.202503462
- Haq IU, Ahmad I, Shabbir MA, et al. Using immunoinformatics to design a rational in silico vaccine against human astrovirus targeting capsid polyprotein VP90. In Silico Pharmacol. 2025;13(3):e21.
- Khan Z, Ahmad I, Shabbir MA, et al. In silico multi-epitope vaccine candidate against type 1 parainfluenza virus. Comput Biol Med. 2023;154:e106601. https://doi.org/10.21203/rs.3.rs-2455059/v1
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for recognition of errors in three-dimensional protein structures. Nucleic Acids Res. 2007;35:W407-W410. https://doi.org/10.1093/nar/gkm290
- Sippl MJ, Lackner P, Domingues FS, et al. Assessment of the CASP4 fold recognition category. Proteins. 2001;45(S5):55-67. https://doi.org/10.1002/prot.10006
- Khan B, Ahmad I, Haq IU, et al. Systematic identification of molecular biomarkers and drug candidates targeting MAPK3 in multiple sclerosis. Hum Gene. 2025;12:e201436. https://doi.org/10.1016/j.humgen.2025.201436
- Rahiyab M, Ahmad I, Shabbir MA, et al. Computational profiling of molecular biomarkers in congenital disorders of glycosylation type I and binding analysis of ginkgolide A with P4HB. Comput Biol Med. 2025;190:e110042. https://doi.org/10.1016/j.compbiomed.2025.110042
- Shey RA, Ghogomu SM, Esoh KK, et al. In silico design of a multi-epitope vaccine candidate against onchocerciasis. Sci Rep. 2019;9(1):e4409. https://doi.org/10.1038/s41598-019-40833-x
- Alizadeh M, Amini-Khoei H, Ahmad I, et al. Designing a novel multi-epitope vaccine against Ebola virus using reverse vaccinology. Sci Rep. 2022;12(1):e7757. https://doi.org/10.1038/s41598-022-11851-z
- Ponomarenko J, Bui HH, Li W, et al. ElliPro: a new structure-based tool for prediction of antibody epitopes. BMC Bioinformatics. 2008;9:e514. https://doi.org/10.1186/1471-2105-9-514
- Xagorari A, Chlichlia K. Toll-like receptors and viruses: induction of innate antiviral immune responses. Open Microbiol J. 2008;2:49-59. https://doi.org/10.2174/1874285800802010049
- Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein–protein docking. Nat Protoc. 2020;15(5):1829-1852. https://doi.org/10.1038/s41596-020-0312-x
- Ahmad S, Ahmad I, Haq IU, et al. Exploring the potential mechanism of Tinospora cordifolia in cancer treatment using network pharmacology and molecular docking. In Silico Res Biomed. 2025;4:e100124.
- López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42(Web Server issue):W271-W276. https://doi.org/10.1093/nar/gku339
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16(6):276-277. https://doi.org/10.1016/S0168-9525(00)02024-2
- Ahmad S, Haq IU, Shabbir MA, et al. Design of a novel multi-epitope vaccine against human torovirus disease using immunoinformatics. Comput Biol Chem. 2024;113:e108213. https://doi.org/10.1016/j.compbiolchem.2024.108213
- Grote A, Hiller K, Scheer M, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33:W526-W531. https://doi.org/10.1093/nar/gki376
- Samad A, Ahmad I, Haq IU, et al. Designing a multi-epitope vaccine against SARS-CoV-2 using an immunoinformatics approach. J Biomol Struct Dyn. 2022;40(1):14-30.
- Khan S, Ahmad I, Haq IU, et al. Exploring stevioside binding affinity with proteins involved in diabetic signaling pathways: an in silico approach. Front Pharmacol. 2024;15:e1377916. https://doi.org/10.3389/fphar.2024.1377916
- Abdi SAH, Ahmad I, Haq IU, et al. Multi-epitope-based vaccine candidate for monkeypox virus: an in silico approach. Vaccines. 2022;10(9):1564. https://doi.org/10.3390/vaccines10091564
- Souod N, Ahmad I, Haq IU, et al. In silico design and evaluation of a multiepitope vaccine against Bordetella pertussis. Genomics Inform. 2025;23:e16.
- Sethi G, Ahmad I, Haq IU, et al. Designing a broad-spectrum multi-epitope subunit vaccine against leptospirosis using immunoinformatics. Front Immunol. 2025;15:e1503853. https://doi.org/10.3389/fimmu.2024.1503853
- Yang Z, Bogdan P, Nazarian S. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci Rep. 2021;11(1):e3238. https://doi.org/10.1038/s41598-021-81749-9
- Khan S, Ahmad I, Haq IU, et al. Developing a novel computational strategy for a multi-epitope vaccine against Guanarito virus. J Emerg Trends Novel Res. 2024;11(1):1-12.
- Foroogh N, Ahmad I, Haq IU, et al. Structural and functional characterization of the FimH adhesin of uropathogenic Escherichia coli. J Struct Biol. 2021;161:e105288. https://doi.org/10.1016/j.micpath.2021.105288
- Narula A, Pandey RK, Khatoon N, et al. Excavating chikungunya genome to design B- and T-cell multi-epitope subunit vaccine using immunoinformatics. Infect Genet Evol. 2018;61:4-15. https://doi.org/10.1016/j.meegid.2018.02.001
- Alizadeh M, Ahmad I, Haq IU, et al. Designing a novel multi-epitope vaccine against Ebola virus using reverse vaccinology. Sci Rep. 2022;12(1):e7757.
- Roundy CM, Azar SR, Rossi SL, Weaver SC. Insect-specific viruses: a historical overview and recent developments. Adv Virus Res. 2017;98:119-146. https://doi.org/10.1016/bs.aivir.2016.10.001
- Bettis AA, Jackson ML, Yoon IK, et al. The global epidemiology of chikungunya from 1999 to 2020. PLoS Negl Trop Dis. 2022;16(1):e0010069. https://doi.org/10.1371/journal.pntd.0010069
- Hierlihy C, Zambrano H, Alarcon-Elbal PM, et al. Individual and community mitigation measures for chikungunya virus prevention: a systematic review. PLoS One. 2019;14(2):e0212054. https://doi.org/10.1371/journal.pone.0212054
- Romano P, Giugno R, Pulvirenti A. Tools and collaborative environments for bioinformatics research. Brief Bioinform. 2011;12(6):549-561. https://doi.org/10.1093/bib/bbr055
- Robson B, Boray S. Web-based universal exchange and inference language for medicine. Comput Biol Med. 2015;66:82-102. https://doi.org/10.1016/j.compbiomed.2015.07.015
- An W, Liu Y, Wang X, et al. Recent progress on chikungunya virus research. Virol Sin. 2017;32:441-453. https://doi.org/10.1007/s12250-017-4072-x
- Fros JJ, van der Maten E, Vlak JM, Pijlman GP. Chikungunya virus nsP2 inhibits type I/II interferon-stimulated JAK–STAT signaling. J Virol. 2010;84(20):10877-10887. https://doi.org/10.1128/JVI.00949-10
- Bae S, Lee JY, Myoung J. Chikungunya virus proteins nsP2, E2, and E1 antagonize interferon-β signaling. Viruses. 2019;11(7):e584. https://doi.org/10.3390/v11070584
- Göertz GP, McNally KL, Robertson SJ, et al. The methyltransferase-like domain of chikungunya virus nsP2 inhibits interferon response. J Virol. 2018;92(17):e01008-18. https://doi.org/10.1128/JVI.01008-18
- Schrauf S, Tschismarov R, Tauber E, et al. Current efforts in vaccine development for Zika and chikungunya virus infections. Front Immunol. 2020;11:e592. https://doi.org/10.3389/fimmu.2020.00592
- Khan A, Ahmad I, Haq IU, et al. Allosteric ligands targeting flavivirus NS5 identified from ZINC database. J Mol Graph Model. 2018;82:37-47.
- Pandey RK, Sundar S, Prajapati VK. Differential expression of miRNAs regulates T-cell differentiation during visceral leishmaniasis. Front Microbiol. 2016;7:206. https://doi.org/10.3389/fmicb.2016.00206
- Aslam S, Ahmad I, Haq IU, et al. Designing a multi-epitope vaccine against Chlamydia trachomatis. Biology (Basel). 2021;10(10):e997. https://doi.org/10.3390/biology10100997
- Mahmud S, Ahmad I, Haq IU, et al. Designing a multi-epitope vaccine candidate against MERS-CoV. Sci Rep. 2021;11(1):e15431.
- Kadam A, Sasidharan S, Saudagar P. Computational design of a multi-epitope vaccine against Ebola virus. Infect Genet Evol. 2020;85:e104464. https://doi.org/10.1016/j.meegid.2020.104464
- Shankar U, Gupta A, Kumar R, et al. Mining Ebola virus genome for multi-epitope vaccine construction. J Biomol Struct Dyn. 2022;40(11):4815-4831.
- Qamar MT, Ahmad S, Fatima I, et al. Designing a multi-epitope vaccine against Staphylococcus aureus. Comput Biol Med. 2021;132:e104389. https://doi.org/10.1016/j.compbiolchem.2021.104389
- Solanki V, Tiwari V. Subtractive proteomics and reverse vaccinology for chimeric vaccine design against Acinetobacter baumannii. Sci Rep. 2018;8(1):e9044.
- Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, ed. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005:571-607.
- Hussain I, Ahmad I, Haq IU, et al. Structure-guided drug repurposing reveals antiviral candidates for Bourbon virus. In Silico Pharmacol. 2025;13(3):155. https://doi.org/10.1007/s40203-025-00443-0
- Khan S, Ahmad I, Haq IU, et al. Design of a novel multi-epitope vaccine against Bundibugyo ebolavirus. Med Omics. 2025;8:e100050. https://doi.org/10.1016/j.medomic.2025.100050
- Morla S, Makhija A, Kumar S. Synonymous codon usage patterns in rabies virus glycoprotein gene. Gene. 2016;584(1):1-6. https://doi.org/10.1016/j.gene.2016.02.047
- Brinton MA. The molecular biology of West Nile virus. Annu Rev Microbiol. 2002;56:371-402.
- Kar T, Narsaria U, Basak S, et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep. 2020;10(1):e10895. https://doi.org/10.1038/s41598-020-67749-1
- Safavi A, Kefayat A, Mahdevar E, et al. Exploring hidden antigens of SARS-CoV-2 to design a multi-epitope vaccine. Vaccine. 2020;38(48):7612-7628. https://doi.org/10.1016/j.vaccine.2020.10.016